This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDAApproves Merck’s ENFLONSIA™ to Protect Infants from Severe RSV Illness Merck operating as MSD outside the United States and Canada, has received a significant regulatory milestone with the U.S. Food and Drug Administration (FDA) granting approval for ENFLONSIA™ (clesrovimab-cfor).
The goal is to boost SMN protein production more effectively and offer a more convenient, once-a-year dosing schedule , thereby improving long-term disease management and patient compliance. These reductions were sustained through one year , highlighting salanersen’s potential to meaningfully slow disease progression. Source link
Despite recent advances in gene therapy for sickle cell disease (SCD) , automated red blood cell exchange (aRBCX) remains a cornerstone therapy that plays a vital yet underutilised role in managing complications and enhancing quality of life for millions living with this devastating condition worldwide.
Xywav: A Low-Sodium Alternative with FDAApproval Xywav is a uniquely formulated, low-sodium oxybate therapy, and remains the only product of its kind approved by the U.S. It is also approved for adult patients with idiopathic hypersomnia (IH). mmHg (P=0.0003) In-office seated resting SBP decreased by −9.2
Over 70% of new patients are diagnosed with stage IV disease, also called extensive stage disease (ES-SCLC). The results were remarkable for this very sick patient population, with a median OS of 14 months at the dose of 10 mg given every 2 weeks: The data supported FDAapproval, in May 2024, of tarlatamab for refractory ES-SCLC.
Primary membranous nephropathy is a rare and serious autoimmune kidney disease characterized by the formation of autoantibodies that damage the glomerular basement membrane, often resulting in nephrotic syndrome —a condition marked by high levels of protein in the urine, severe swelling, and a significant risk of progression to kidney failure.
The study, which is the largest to date that focuses exclusively on this patient population, shows high overall response rates (ORR) alongside deep and durable responses, offering much-needed hope for individuals whose disease typically carries a poor prognosis. Head of Myeloma Unit at Tel-Aviv Sourasky Medical Center in Israel.
Food and Drug Administration (FDA)-approved gene therapy for the treatment of Duchenne muscular dystrophy (DMD). The ultimate aim is to enable the administration of ELEVIDYS safely while honoring its potential to provide a dramatic and durable therapeutic benefit for children battling this profoundly challenging disease.
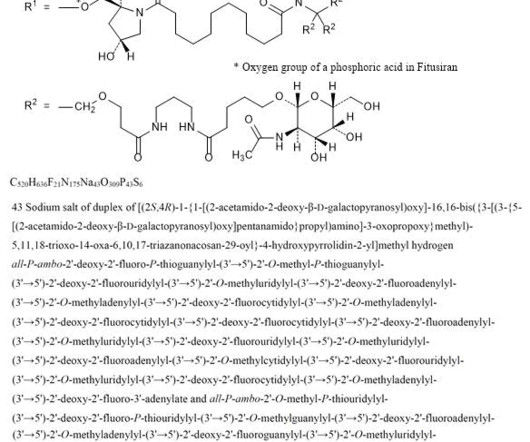
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 1] [2] Adverse effects The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal). [2]
This trial was the first in its therapeutic area to use a composite primary endpoint consisting solely of major morbidity and mortality events , such as all-cause death, lung transplantation, or PAH-related hospitalization of at least 24 hours. WINREVAIR is the first FDA-approved activin signaling inhibitor for PAH.
FDAApproves KEYTRUDA® (Pembrolizumab) for Perioperative Treatment of Resectable Locally Advanced Head and Neck Squamous Cell Carcinoma Merck known as MSD outside the United States and Canada, recently announced that the U.S. Among these patients, the median event-free survival was 59.7 0.89; p = 0.00140). 0.89; p = 0.00140).
Deep Dive Library Events Press Releases Topics Sign up Search Sign up Search Pharma Biotech FDA Clinical Trials Deals Drug Pricing Gene Therapy An article from Dive Brief Moderna COVID vaccine gets full approval for children The approval comes amid regulatory upheaval under HHS head Robert F. Kennedy Jr. Kennedy Jr.
FDA Expands Approval of AbbVie’s MAVYRET® as First and Only 8-Week Treatment for Acute Hepatitis C in Adults and Children Aged 3 and Above AbbVie has received a significant regulatory boost for its hepatitis C treatment portfolio as the U.S. Global and U.S. Safety outcomes from the trial further bolster the confidence in MAVYRET’s use.
A new drug has entered the arsenal against Duchenne muscular dystrophy (DMD), a genetic disease that affects boys and is challenging to treat. FDA classifies it as a “nonsteroidal treatment” – not a gene therapy, but it affects gene expression. ITF Therapeutics provides the new drug in the US. Hyperbole or Hope?
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. This indication is approved under accelerated approval regulation based on overall response rate and duration of response. This indication is approved under accelerated approval based on overall response rate and duration of response.
CB1 and CB2 agonists exhibit broad anti-inflammatory properties, suggesting their potential to treat inflammatory diseases. Secondary VAS and pharmacokinetic (PK) endpoints and adverse events were assessed. Drug Liking and all other VAS outcomes were greatest for nabilone 3mg and 6mg, which is a currently FDA-approved medication.
FDAApproves Imcivree (setmelanotide) for Chronic Weight Management in Patients with Obesity Due to POMC, PCSK1 or LEPR Deficiency. Nasdaq:RYTM), a biopharmaceutical company aimed at developing and commercializing therapies for the treatment of rare genetic diseases of obesity, announced today that the U.S. BOSTON, Nov.
FDAApproves Veklury (remdesivir) for the Treatment of COVID-19. Food and Drug Administration (FDA) has approved the antiviral drug Veklury (remdesivir) for the treatment of patients with COVID-19 requiring hospitalization. The incidence of adverse events associated with Veklury was similar to placebo in the ACTT-1 trial.
Novartis today announced the US Food and Drug Administration (FDA) has granted accelerated approval for Kymriah ® (tisagenlecleucel) for the treatment of adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two or more lines of systemic therapy.
FDAapproves Pfizer’s LITFULO™ (ritlecitinib) for adults and adolescents with severe alopecia areata Pfizer Inc. Food and Drug Administration (FDA) has approved LITFULO™ (ritlecitinib), a once-daily oral treatment, for individuals 12 years of age and older with severe alopecia areata. with placebo.
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. Food and Drug Administration (FDA) has approved Klisyri (tirbanibulin) for the topical treatment of actinic keratosis (AK) on the face or scalp. The FDAapproval of Klisyri is a significant milestone for Athenex.
Clinical trials for ultra-rare diseases can be particularly challenging to mount due to small, geographically-dispersed patient populations. For such trials, the US Food and Drug Administration (FDA) may allow the use of credible real-world data (RWD) and real-world evidence (RWE) in lieu of data collected in a Phase 3 trial.
”
As previously announced, the planned trial will evaluate the efficacy of Berubicin in patients with GBM who have failed primary treatment for their disease. In this trial the overall response rate of stable disease or better was 44%. These statements relate to future events, future expectations, plans and prospects.
1 High levels of LDL-C starting at birth accelerate the event of atherosclerotic disorder , resulting in an overall increased risk of cardiovascular events, including attack and other vascular conditions, at an earlier age.2 HeFH is an inherited, genetic condition with a prevalence of 1 in 250 people worldwide.1
RAD011 is a pivotal-trial ready synthetic cannabidiol oral solution with potential utilization in multiple endocrine and metabolic orphan diseases.
Prader-Willi syndrome (“PWS”) will be the initial indication, which has been granted Orphan Drug and Fast Track Designation by the FDA.
Disease Highlights.
Food and Drug Administration (FDA) has now approved the very first oral drug to treat the millions of men affected. . However, Myovant’s drug dropped the percentage of major cardiovascular events to 2.9% compared to AbbVie’s at 6.2%. This should make the drug more appealing to prescribing physicians. .
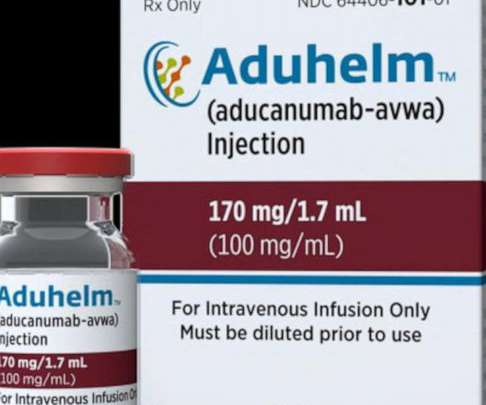
ADUHELM should be initiated in patients with mild cognitive impairment due to Alzheimer’s disease or mild Alzheimer’s dementia. Food and Drug Administration (FDA) has approved an updated label for ADUHELM (aducanumab-avwa) injection 100 mg/mL solution. ADUHELM is indicated for the treatment of Alzheimer’s disease.
Food and Drug Administration (FDA) approved Actemra ® /RoActemra ® (tocilizumab) subcutaneous injection for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD), a debilitating condition with limited treatment options.
Pulmonary arterial hypertension (PAH) is a progressive and fatal lung disease that is caused or influenced by multiple factors. As your CRO partner, we deliver equally dedicated and experienced teams that understand this disease and the range of assessments used for collecting endpoint data in PAH clinical trials.
Without treatment, infants do not achieve these milestones in the natural history of the disease. There were no treatment-related adverse events leading to withdrawal. Evrysdi is currently approved in 81 countries and the dossier is under review in a further 27 countries.
The Pfizer-BioNTech COVID-19 Vaccine has not been approved or licensed by the U.S. Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) for use in individuals 16 years of age and older.
(NYSE American:PLX) (TASE:PLX), a biopharmaceutical company focused on the development, production and commercialization of recombinant therapeutic proteins produced by its proprietary ProCellEx ® plant cell-based protein expression system, and Chiesi Global Rare Diseases , a business unit of Chiesi Farmaceutici S.p.A.,
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of LN. ABOUT AURINIA.
” In this head-to-head Phase IV study, patients in the Aimovig arm demonstrated a significantly lower discontinuation rate due to adverse events versus patients in the topiramate arm (10.6% Additional study treatment-related adverse events reported by ?2% versus 38.9%). .” versus 38.9%). versus 31.2%).
We believe Category I status for the procedure of inserting a drug eluting insert into the lacrimal canaliculus delivers immediate benefits for not only DEXTENZA, but also for the other product candidates in our pipeline portfolio that are designed to utilize the same route of administration, including our two programs in dry eye disease.”.
EST.
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of lupus nephritis (LN).
Teva and MedinCell Announce FDAApproval of UZEDY™ (risperidone) Extended-Release Injectable Suspension, a Long-Acting Subcutaneous Atypical Antipsychotic Injection, for the Treatment of Schizophrenia in Adults Teva Pharmaceuticals, a U.S. The primary endpoint was the frequency of all adverse events, including serious adverse events.
Food and Drug Administration (FDA) has accepted the company’s supplemental New Drug Application (sNDA) and granted Priority Review for Esbriet ® (pirfenidone) for the treatment of unclassifiable interstitial lung disease (UILD). The FDA is expected to make a decision on approval by May 2021. Since its U.S. mL, p=0.002).
Overall, the number of treatment-emergent adverse events reported was similar between the ponesimod and teriflunomide treated groups, and the majority were mild/moderate and did not warrant treatment discontinuation. Adverse events should be reported. vs. 9.4%), nasopharyngitis (19.3% vs. 16.8%), headache (11.5% vs. 10.4%).
People with type 2 diabetes are at increased risk for heart attacks, stroke, and other forms of cardiovascular disease, and at an earlier age than other people. In fact, the evidence suggests that such drugs might even offer some protection against heart disease.
BYOOVIZ™ is the first FDAapproved ophthalmology biosimilar BYOOVIZ, priced 40% lower than LUCENTIS®, provides an equally effective and more affordable treatment option to patients suffering from retinal disorders BYOOVIZ will be commercially available through major distributors across the U.S. on July 1, 2022. Biogen Inc.
In the initial 275 patients, rates of adverse events (AEs) were similar among groups. Authorized Emergency Use Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use.
.
Moleculin recently announced that the FDA had allowed its request for Investigational New Drug (IND) status for Annamycin, allowing Moleculin to begin a Phase 1B /2 clinical trial in the US for patients with soft tissue sarcoma (STS) that has metastasized to the lungs after first-line therapy for their disease.
If approved, Xarelto will be the only oral Factor Xa Inhibitor indicated in the U.S. There are currently no FDA-approved anticoagulation therapies for pediatric patients with congenital heart disease who have undergone the Fontan procedure. for use in pediatric patients. EINSTEIN-Jr. About the EINSTEIN-Jr.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content