This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Deep Dive Library Events Press Releases Topics Sign up Search Sign up Search Pharma Biotech FDA Clinical Trials Deals Drug Pricing Gene Therapy An article from Dive Brief Actithera draws new investors to radiopharma drug pitch The four-year-old biotech raised about $75 million in a Series A round that involved nine venture capital firms.
The 2025 CDD User Group Meeting (UGM) Canada brought together researchers, industry experts, and thought leaders to discuss the latest advancements in drug discovery, data management, and collaborative research.
Given macro healthcare influences (eg, economic uncertainty, environmental changes) and the numerous available treatments for major diseases, drug developers may need to reassess their therapeutic strategies. This has led drug developers to unintentionally limit their potential within chosen therapeutic spaces.
Drug Discovery 101 – Webinar Recording Starting a career in drug discovery can feel overwhelming, but breaking down the process makes it more approachable. Our Drug Discovery 101 webinar outlines the entire process from start to finish. The Challenges Ahead Discovering drugs isn’t a walk in the park.
Haemoglobin A1c (HbA1c) is a validated surrogate endpoint for the reduction of microvascular complications associated with diabetes mellitus; reduced HIV-RNA levels serve as an endpoint for HIV disease control; and a reduction in low-density lipoprotein (LDL) cholesterol is used as an endpoint indicating lower likelihood of cardiovascular events.
By Sarah Wicks & Dara Katcher Levy FDAs Office of Prescription Drug Promotion (OPDP) issued its second Untitled Letter of 2025 to Taiho Oncology (Taiho) for a healthcare provider branded website for its drug LYTGOBI (futibatinib). Join us as we break down what went wrong this time. What Happened? The problem?
Antibody-drugconjugates (ADCs) represent a significant advancement in drug discovery, combining the precision of monoclonal antibodies with the cancer-killing power of cytotoxic drugs. This type of analysis is crucial for drug developers, providing a framework to identify and mitigate toxicity risks early in the design process.
Safety: Diarrhea was the most frequent adverse event, but serious adverse events were balanced across treatment groups. Not Yet Approved: Nerandomilast is still an investigational drug and is not yet approved for use. FDA Designation: Nerandomilast received Breakthrough Therapy Designation from the FDA for the treatment of IPF.
The challenge of GPCR drug discovery G protein-coupled receptors (GPCRs) are one of the most desirable and challenging target classes in drug discovery, as their mutation can lead to a wide range of diseases such as cancer, cardiovascular disorders and neurological conditions.
According to Merck, the addition of WINREVAIR to background therapy resulted in a meaningful improvement in patient outcomes, particularly in delaying disease progression and reducing the risk of severe clinical events. These data underscore the therapeutic potential of WINREVAIR when introduced early in the course of disease.
Unlike most approved drugs, which work by turning cell signaling up or down like a dimmer switch, this approach aims to rid cells of problem proteins entirely. Earlier this year, the field got its best look yet at how well one common type of these drugs performs. Several have drugs in or near clinical testing.
Most adverse events (AEs) were mild to moderate in severity. Biogen emphasized that no dose-limiting toxicities were observed, and there were no reports of serious adverse events directly attributable to the study drug. The most common AEs included pyrexia (fever) and upper respiratory tract infections.
Crucially, weight loss in the MariTide-treated groups had not plateaued at the 52-week mark , indicating the drug’s potential to drive continued weight reduction beyond one year. Safety Profile and Tolerability: No Surprises, GI Events Mostly Mild In terms of safety, the Phase 2 study did not identify any new safety signals.
One area drawing increasing attention is how these changes will affect the drug development and review process. As reported by the Wall Street Journal last week , reviews of both innovative and follow-on drugs have been caught in a traffic jam, largely due to a leadership vacuum at the Center for Drug Evaluation and Research (CDER).
Altasciences At CPHI Americas 2025 pmjackson Thu, 07/03/2025 - 13:41 In the ever-changing drug development world, staying agile is essential to enabling more informed decisions, faster. It’s a challenge that affects a large percentage of new drug candidates—one we’re passionate about helping to solve.
There were no thromboembolic events, hypersensitivity reactions, or treatment-emergent adverse events (TEAEs) that led to discontinuation. Strong Safety Profile Reinforces Mim8’s Potential Mim8’s safety profile was a central focus of the FRONTIER5 trial.
From foundational multimodal models to targeted disease prediction, phenotyping, and drug repurposing, ML4H combines large-scale data, deep clinical insight, and cutting-edge AI methods to drive innovation across cardiology, neurology, and beyond.
Revolutionizing Generic Drug Development: The Power of Digital Transformation As the pharmaceutical industry continues to evolve, one thing is clear: digital transformation is no longer a luxury, but a necessity. For years, generic drug manufacturers have relied on traditional methods to develop and launch new products.
Misrepresentation of Drugs in the Unregulated Market "The misrepresentation of illicit drugs is a persistent problem in unregulated markets ( Barratt et al., When users consume illicit drugs of unknown content, quality, and dosage, their risk of overdose and other adverse health events increases significantly ( Singh et al.,
The Unseen Heroes of Generic Drug Development: Pharmacovigilance As a generic drug developer, you're no stranger to the challenges of bringing affordable medications to market. But have you ever stopped to think about the unsung heroes of generic drug development? Share your experiences and insights in the comments below!
These applications are incredibly useful for basic research, studying the environment , probing different aspects of biotechnology , or contributing to undertakings like drug discovery. Improved understanding Understanding metabolism is also crucial for drug discovery and investigating disease mechanisms. coli enzyme (Fig.
Food and Drug Administration (FDA) for the treatment of cataplexy or excessive daytime sleepiness (EDS) in patients aged seven years and older with narcolepsy. of participants reported treatment-emergent adverse events (TEAEs), all of which were mild or moderate and consistent with previous safety data on Xywav. Approximately 32.8%
Network like a boss at Drug Discovery 2025 If you’re early in your career, there’s a good chance you’ve been told to “build your network”. Often described as a professional speed dating session, it’s a fast, informal way to meet people working across the drug discovery field. Or where to start. Network Like a Boss is our answer.
The most frequently reported treatment-emergent adverse events (reported by greater than 10% of patients) included diarrhea, COVID-19, dizziness, dry mouth, and nausea — all of which were predominantly mild to moderate in severity. Fast Track and Orphan Drug Designations from FDA The U.S.
The most common adverse events were mild to moderate in intensity and did not differ significantly from known safety patterns. In both the European Union (EU) and the United States , the drug is currently approved as an add-on therapy for seizures associated with Dravet syndrome and Lennox-Gastaut syndrome in patients aged 2 years and older.
Let’s dive in… Wave 2: B7-H3 Optimistically named B7-homologue-3, B7-H3, a distant relative of the immune regulatory proteins B7-1 and B7-2, has recently moved into the spotlight - not as an immune regulator but as a target of advanced therapeutic modalities including bispecific antibodies, antibody drug conjugates (ADC) and CAR-T cells.
As a proof of concept, a drug designed to target a newly discovered biological node is showing efficacy in treating rare genetic diseases in the kidney, the eye, and the brain and is now making its way to clinical trials in collaboration with a pharmaceutical partner. We will keep you in the loop on upcoming events and research highlights.
Published July 14, 2025 Ben Fidler Senior Editor post share post print email license Takeda reported positive results for oveporexton, a drug being developed for narcolepsy Type 1, on July 14, 2025. The drug, formerly known as TAK-861 but now called oveporexton, was evaluated in two Phase 3 studies in a main type of narcolepsy.
The double-blind treatment period lasted 24 weeks, during which both efficacy and adverse events (AEs) were closely monitored. Primary Endpoint Met: Improved Tolerability with Atogepant At the core of the TEMPLE study was the investigation of treatment discontinuation rates due to adverse events, a key measure of real-world drug tolerability.
Food and Drug Administration (FDA) as a monotherapy for children with achondroplasia. Safety and Tolerability Safety remains a top priority in pediatric drug development, and the interim results offer reassurance. Administered once weekly, TransCon CNP is currently under Priority Review by the U.S.
How will the imposition of tariffs and a burgeoning trade war affect domestic drug production and companies supply chain demands? The answer to that is still no, the current state of world and governmental events notwithstanding. Also, to be sure, quality still matters in the drug and device industries.
Achieving the Primary Endpoint: Sotyktu Demonstrates Superiority Over Placebo The POETYK PsA-1 trial, officially designated as study IM011-054, enrolled patients with active PsA who had not received prior treatment with biologic disease-modifying antirheumatic drugs (bDMARDs).
Now to antibody-drug conjugates (ADC) where we saw some early programs with difficult toxicities. 4 patients (33%) had grade 4 adverse events. The Next Wave: Targeting B7H3 – a drug development tsunami in the making? That is a decent hypothesis to explore and their early clinical data at ASCO 2025 looked interesting. Stay tuned.
Prof Rory Johnson, Associate Professor, University College Dublin, and Dr Shalini Andersson, Vice President Nucleic Acid Therapeutics, AstraZeneca will lead this years event focussed on drugging the undruggable.
As a result of reforms enacted by the Food and Drug Omnibus Reform Act of 2022 (FDORA), FDAs authority to approve as well as withdraw an accelerated approval was given new regulatory teeth to ensure that confirmatory trials are conducted expeditiously. However, the meaning of due diligence has long been a question.
Food and Drug Administration (FDA)-approved gene therapy for the treatment of Duchenne muscular dystrophy (DMD). ELEVIDYS is currently the only U.S. The company’s updated guidance focuses on strengthening its safety profile in non-ambulatory patients following a second report of acute liver failure (ALF) that resulted in a patient’s death.
Drug development is a complex and highly regulated process. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and other global counterparts, set rigorous standards to ensure that drugs are safe, effective, and high-quality. Regulatory agencies, such as the U.S.
Sponsors find that an FSP solution is often the best choice to help advance their drug development projects, whether they need to fill small gaps in services or support large-scale programs with dedicated teams across functions. Central to this framework is the concept of risk-based monitoring (RBM).
Food and Drug Administration (FDA) has granted Fast Track Designation (FTD) to efgartigimod for the treatment of primary Sjogren’s disease, further accelerating its pathway toward potential regulatory approval. Additionally, argenx announced that the U.S. No majority of placebo patients reached this threshold during the study.
Treatment-emergent adverse events (TEAEs) occurred in 76.3% Food and Drug Administration (FDA) granted the therapy Priority Review status in May 2025 following its Biologics License Application (BLA) submission in March, reflecting the agency’s recognition of its potential to address a serious condition with limited treatment options.
Safety and Tolerability The safety profile of MR-141 in VEGA-3 was consistent with earlier clinical trials of the drug. No new safety signals were identified, and there were no treatment-related serious adverse events reported during the study.
Continued authorization of Ziihera for this indication will depend on positive outcomes from the ongoing Phase 3 HERIZON-BTC-302 trial , which is evaluating the drug in combination with standard-of-care therapy in the first-line setting. of patients, with the most frequent being diarrhea, fatigue, and elevated liver enzymes. In the U.S.,
Additionally, the tablet form supports flexible dosing and simplifies dose reductions, which may be required due to drug interactions or adverse events, as guided by the product’s label. Food and Drug Administration (FDA) granted approval to the tablet formulation of BRUKINSA for all five of its approved indications.
The pharmaceutical industry faces a persistent challenge: despite significant investments in drug development, a substantial proportion of promising candidates fail due to unforeseen toxicity issues.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








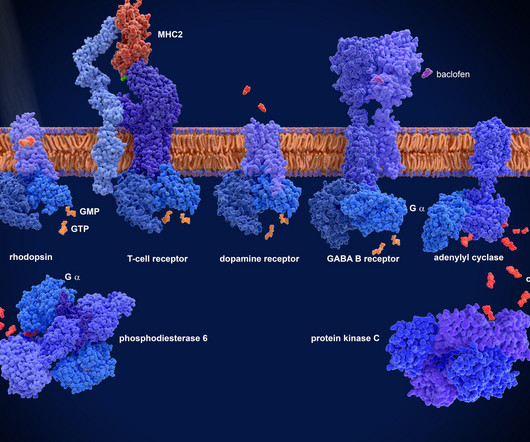







































Let's personalize your content