This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Accelerated Approval of Drugs and Biologics Administrative/ Procedural New Civil Monetary Penalties for Failure to Meet Accelerated Post Marketing Requirements Administrative/ Procedural Carried over from previous guidance agenda Exclusivity for First Interchangeable Biosimilar Biological Products Administrative/ Procedural Carried over from previous (..)
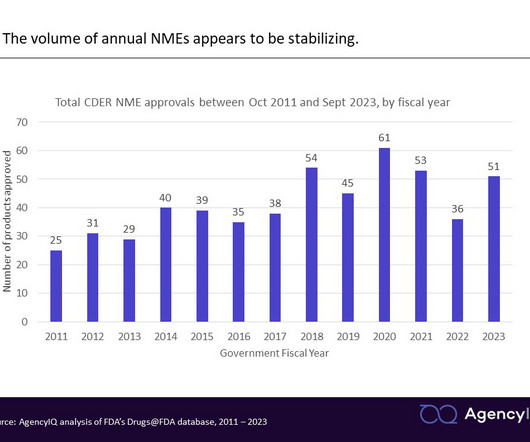
CDER is the FDA office in charge of reviewing pharmaceuticals and therapeutic biologics. All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics). The FDA also refers to novel products as “New Molecular Entities,” or NMEs.
fit in this definition. The FDA DSUR is intended to be consistent with the format and content for IND annual reporting supported by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and is more comprehensive, and informative than the IND annual report currently required by FDA.
12/29/2023 FDORA, Section 3602 Clinical Trials Modernization : FDA is directed to require the submission of a “diversity action plan” for all Phase 3 clinical trials of new drugs. fit in this definition. ” The report will also cover Federal agency roles in addressing vulnerabilities and statutory limitations.
Proposed Rule Rulemaking to Provide by Regulation that an Ingredient Is Not Excluded From the Dietary Supplement Definition December 2024 This proposed rule, if finalized, would allow a specific ingredient to be marketed in or as a dietary supplement. and the E.C.
To this end, FDA recently announced the publication of a guidance titled “Considerations for Long-Term Clinical Neurodevelopmental Safety Studies in Neonatal Product Development,” (the “Guidance”). This guidance finalizes a draft guidance issued in February 2023.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













Let's personalize your content