This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
In this article, we will delve into the benefits of working with integrated CDMO services and explore how they can streamline the drug development and manufacturing process. The Rise of Integrated CDMOs The global biotechnology and pharmaceutical services outsourcing market size was valued at $70.48 from 2023 to 2030.
This article aims to provide a comprehensive overview of the key aspects of the regulatory framework, highlighting the requirements and challenges faced by pharmaceutical companies seeking to introduce generic drugs into the Japanese market. New Drug Application (NDA) : Needed for marketing approval of new drugs.
Navigating the Complex World of Global Drug Patents: Strategies and Challenges Ahead As a pharmaceutical professional, you know how crucial it is to protect your innovative drug patents in the global market. But what can you do to protect your drug patent in global markets? Read the full article here: [link]
These innovations have started to shift industry perceptions, positioning AI as a transformative tool that could alter how drugs are developed, tested, and brought to market. Regulators care a lot about controlling the Type 1 error rate of the clinical trial, Smith notes.
Unlearn’s early collaboration with regulators has helped it navigate this space effectively. His expertise spans business development, strategic planning, product management and marketing, with a focus on sustainable growth and portfolio expansion.
For example, transcriptomic processes are showing the potential to identify and track failures in gene expression and gene regulation of amyloid and tau-related biomarkers, understood as precursors to the onset of Alzheimers disease (AD).
The global market for CNS therapeutics was worth an estimated $144.3 Developers and sponsors working on biologically derived therapies in the US can utilise the Regenerative Medicine Advanced Therapy (RMAT) designation, which regulators grant to promising regenerative therapies. Pandit R, Chen L, Götz J. Adv Drug Deliv Rev.
Challenge #4: Regulatory and market access hurdles The regulatory and health technology assessment (HTA) pathways for rare disease therapies are complex and vary by region. With the new EU HTA Regulation impacting orphan medicines, navigating these pathways has become even more challenging.
Faster time-to-market and reduced costs. By integrating AI into the manufacturing process, the developer achieved higher consistency across batches, improved overall product quality, and reduced time-to-market – all critical factors in ensuring patients receive timely and effective treatment. The result?
In this first article of the two-part series, we cover the challenges of AI adoption in life sciences drawing lessons from industries that have embraced these tools more rapidly. The challenges lie in regulated industries where governance frameworks for standard quantitative models dont necessarily apply to LLMs.
Lepore highlights strong early progress in oncology, immunology and metabolic diseases, where ProFound has identified novel cytokines, transmembrane tumour antigens and secreted metabolic regulators with significant therapeutic potential. Real, personalised medicine, not just marketing. And the big players are taking notice.
When small and emerging companies are able to build strong, trust-based relationships with regulators, they often see benefits like reduced development risk and accelerated timelines. When teams are well prepared ahead of time, it builds credibility with regulators, and credibility is a valuable currency in the regulatory approval process.
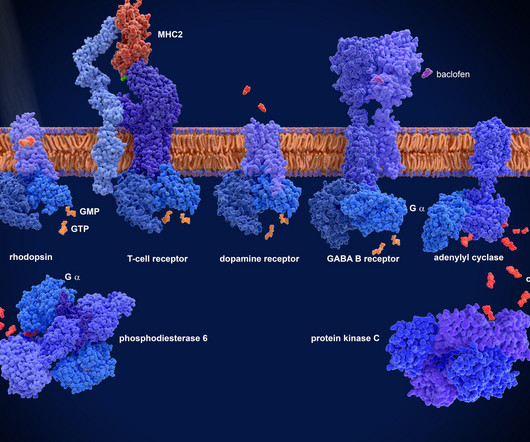
Involved in various physiological processes, such as vision, taste, smell, immune response and neurotransmission, GPCRs are activated by various molecules including hormones, neurotransmitters and environmental stimuli, which trigger a cascade of cellular events that help regulate bodily functions.
However, delays in any of these stages can significantly impact the overall timeline, leading to missed market opportunities and revenue losses. By implementing these strategies, generic drug manufacturers can accelerate their development timelines, improve efficiency, and increase their chances of success in the market.
3-5 These layers of regulation, while essential for animal welfare, make NHP-based studies complex, expensive and time-consuming. However, the last decade has seen a gradual reduction in their use, driven by tightening animal welfare regulations and scientific advances. Are we nearing a post-NHP future?
Multimodality however can detect and connect trends (and in future generate content) across different modalities and therefore allows for better interpretability, which builds trust between regulators, researchers and industry stakeholders. He holds a master’s degree from University of Salerno in political sciences and marketing.
Taking the time to outline broader objectives, such as drug approval, market entry, or expansion into new markets, helps the CRO fully understand your requirements. It’s also important to ask about their past interactions with regulators, including inspections and their outcomes.
At the forefront of this mission is Senior Vice President of Vitalant, Becky Butler Cap, whose career has been long defined by a passion for building new services and treatment options in emerging markets. Building new services and treatment options for patients are a passion for me.
In this article, we'll delve into the world of drafting drug patent applications for biologic drugs. However, the patentability of biologics is governed by specific laws and regulations, such as the Biologics Price Competition and Innovation Act (BPCIA). Read the full article here: [link]
In 2019, we published a detailed peer-review article outlining this process based on our long history of leveraging existing animal and human data to streamline drug development programs and reduce or eliminate animal testing under the 505(b)(2) pathway.[ For questions on NAMs and the impact to your program , contact us.
The antibiotic market has long faced significant barriers to innovation, with structural issues that make it difficult for new antibiotics to gain traction. As the demand for novel antibiotics remains limited due to the preference for cheaper generics, the market has struggled to incentivise much-needed breakthroughs.
Long timelines, strict regulation and scientific uncertainty are part of the job. However, when that pressure is compounded by political shifts, market volatility and rising expectations from the global pharmaceutical industry, the old ways of working start to crack. In early-stage drug discovery, pressure is nothing new.
The Patent Landscape: A Complex Web of Rules and Regulations When it comes to patenting drug combinations, the patent landscape is a complex web of rules and regulations. Here are some tips and tricks from the experts: Conduct thorough market research : Understand the competitive landscape and identify areas of unmet need.
“Regulators are also much more interested in the manufacturing process, especially when it comes to stability.” Because analytical questions from both manufacturers and regulators are increasing, we have to think smarter with novel technologies. We can combine multiple biologically active molecules to create hybrid drugs.
Alamy The Food and Drug Administration will ask Sarepta Therapeutics to halt all shipments of its marketed gene therapy for Duchenne muscular dystrophy, a source familiar with the matter confirmed to BioPharma Dive. You can unsubscribe at anytime. Food and Drug Administration headquarters in Silver Spring, Md.,
Integrating it with external systems—like ERPs, marketing platforms, or data warehouses—is crucial for seamless data exchange. Securing these touchpoints ensures data integrity, confidentiality, and compliance with regulations like GDPR and HIPAA. Check the articles below for more insights. By using OAuth 2.0,
Although not altogether surprising, the formality of an official announcement still came as a shock to many of us who work in areas regulated by HHS, as well as to many others in the public health arena. Gibbs — On Thursday, November 14, President-Elect Trump announced his pick of Robert Kennedy, Jr.,
Dive Insight: Since the FDA’s approval of Novartis’ Kymriah for leukemia in 2017, six more CAR-T therapies have reached market. Alongside removal of the REMS, the regulator also reduced its requirement that patients remain nearby the facility they received treatment from one month to two weeks. You can unsubscribe at anytime.
However, the journey to bringing a generic drug to market is often fraught with challenges. By working closely with partners and regulators, generic drug developers can identify potential issues early on and work together to find solutions. Read the full article here: [link] Share your experiences with us in the comments below!
At Activate and Cyclotron Road, he built fellowships that served as perches for researchers working on societally important ‘ hard’ technologies that might not yet be VC-fundable find their feet and begin developing technologies into market-ready products. Join Asimov Press. It will always be free. I thought, “Wow.
What will the orphan drug market exclusivity haircut mean for industry? Just over a month ago, the European Commission released its proposal for the new pharmaceutical directive and regulation. Fill out the form to read the full article.
Gonzalez — The Wall Street Journal (WSJ) recently published a series of articles as part of its special report “What’s Ahead for Artificial Intelligence.” Three of these articles focus on medical applications of Artificial Intelligence and Machine Learning (AI/ML) and explore FDA’s role in regulating such products.
To kick off 2024, the British device regulator offered its medical device and IVD plans for this year and next, promising public action on the post-market surveillance regulation by mid-2024 and on the core regulations in late 2024 or early 2025. Fill out the form to read the full article.
It appears the issue is largely the result of unclear labeling for biocidal disinfectants, an aspect French regulators may flag in future market authorization evaluations for these products. Regulatory background Biocides are regulated primarily by the Biocidal Products Regulation (BPR) (528/2012/EU).
Commission proposes exemptions to UV-328 ban under POPs Regulation The European Commission plans to implement exemptions introduced under the Stockholm Convention’s 2023 ban of UV-328, an ultraviolet-light absorbing persistent organic pollutant (POP). UV-328 is used in a wide range of applications as a UV absorber and stabilizer.
BY COREY JASEPH, MS, RAC The British medical device regulator just issued its promised framework on international recognition. Fill out the form to read the full article.
I have even heard the phrase “Please check the report, I don’t understand the models and hence trust the number” So, in the risk function, while this is a race for data aggregation, structured data, unstructured data, data quality, data granularity, news feeds, market overviews, its also a challenge from an acceptance perspective.
By Günter Weisshaar The announcement of the In Vitro Diagnostics Regulation (IVDR) in 2017 was celebrated as an essential upgrade to in vitro diagnostic (IVD) device regulations in Europe. The IVDR is the European Union’s regulatory overhaul of pre- and post-market requirements for IVD devices.
The Swiss regulator, Swissmedic, just announced a reorganization, with a new head of the medicinal product authorization and vigilance sector, but also a new and discrete medical devices surveillance sector. Fill out the form to read the full article. AgencyIQ takes a look at what this could mean for medical device manufacturers.
British regulators tease new device regulations in informative live session This week, the British regulator MHRA offered a new peek into its planned medical device regulations. BY COREY JASEPH, MS, RAC | MAR 5, 2024 11:10 PM CST Quick background on medical device regulation in the U.K. post-marketregulation here.]
Swiss regulators align with EU on chemicals, biocides The Swiss Notification Authority for Chemicals has announced new adaptations are in the offing that will harmonize parts of the Swiss Chemicals and Biocidal Products ordinances with recent updates to corresponding EU legislation. the authorization list).
What we expect EU regulators to do in February 2024 Welcome to AgencyIQ’s monthly roundup of EU chemical sector activities. BY SCOTT STEPHENS, MPA | JAN 26, 2024 10:22 PM CST Highlights of upcoming chemical regulatory activities Consultations under the REACH and CLP regulations are coming to an end in February.
What we expect EU regulators to do in December 2023 Welcome to AgencyIQ’s monthly roundup of EU chemical sector activities. For more background on the glyphosate authorization, see AgencyIQ’s October 12 article.] Events happening next month include two ECHA meetings (i.e.,
device regulation timelines To kick off 2024, the British device regulator offered its medical device and IVD plans for this year and next, promising public action on the post-market surveillance regulation by mid-2024 and on the core regulations in late 2024 or early 2025. both pre- and post-market.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.












































Let's personalize your content