This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Biosimilars have emerged as a game-changing force, promising to revolutionize patient access to life-saving biologics while simultaneously reducing healthcare costs. ”[1] The global biosimilarsmarket is experiencing exponential growth, with projections indicating it will reach $69.4 from 2020 to 2025[1].
As demand for these therapies continues to surge, particularly with the emergence of biosimilars, the necessity for strict quality controls during development and manufacturing becomes paramount. These attributes not only determine a drug’s safety and efficacy but also influence its regulatory approval and post-market success.
Claud The ongoing DOGE-led reductions to the federal workforce and recent sweeping policy changes have spawned many questions for compliance officers and quality managers in FDA-regulated companies. If a compliance or quality program exists only as bargaining chip to use with regulators, thats not a formula to instill good habits.
Notably, many of the issues regarding drugs are typically considered the practice of medicine, which FDA does not regulate. Similarly, FDA can use post-marketing requirements (PMRs). That leaves us to speculate what the implications of this Assessment will be.
Unlocking the Potential of Biosimilars: Navigating Regulatory Considerations for Clinical Efficacy Trials As the pharmaceutical industry continues to evolve, biosimilars have emerged as a game-changer in the fight against high healthcare costs. Should the trial compare the biosimilar to the reference product, or to a placebo?
Evaluate the number of products in development, their potential market size, and the competition. Consider the impact of new regulations, such as those related to biosimilars or generic drugs. Regulatory Environment : Changes in regulatory policies can impact a company's valuation.
Global Pharmaceutical Contract Sales Outsourcing Market to Reach $24.8 Billion by 2030: Strategic Insights, Regional Trends, and Technological Shifts Reshaping the Landscape The global pharmaceutical contract sales outsourcing (CSO) market is entering a transformative phase, with its value expected to grow from $17.3 In the U.S.,
For example, many large tech companies market software products with heart rate and other vital signs monitors, and do not ask permission beforehand. It can also provide feedback about a specific product, if the submitter includes information about the intended use, functionality, and desired marketing claims.
Although not altogether surprising, the formality of an official announcement still came as a shock to many of us who work in areas regulated by HHS, as well as to many others in the public health arena. Gibbs — On Thursday, November 14, President-Elect Trump announced his pick of Robert Kennedy, Jr.,
Lenz, Principal Medical Device Regulation Expert & Lisa M. Here, we will describe the recommendations in FDAs guidance documents for collection and processing of data that will be used for training, tuning, and testing AI models and what to include in a marketing submission for AI-enabled software. By Adrienne R.
Livornese As promised in the Fall Unified Regulatory Agenda, FDA issued the final rule to establish the pathway to obtain marketing approval of a nonprescription drug product with an additional condition for nonprescription use (ACNU) on December 26, 2024, before the end of the calendar year. By Deborah L. 105288 (Dec.
Beyond regulatory work, the Commissioner ventured into what he called crossing into advocacy, especially relating to food regulation. Citing the chronic health crisis among children, he challenged attendees to rethink societys tolerance for environmental and dietary toxins, from petroleum-based food dyes to candy coated in talc.
As weve previously blogged, a PCCP is a mechanism established by Congress under the Food and Drug Omnibus Reform Act (FDORA) to streamline post-market changes to medical devices. If FDA agrees to the PCCP, such changes can be made without a supplemental marketing submission.
Gibbs The multi-decade battle over FDAs power to regulate Laboratory Developed Tests (LDTs) had its day in court earlier this week. Many of the fired employees were within the Center for Devices and Radiological Healththe Center that is tasked with regulating all devices, including in vitro diagnostics, which FDA argues includes LDTs.
The biosimilarmarket is finally beginning to fulfill its promise. The latest data show that provider-administered biosimilar drugs are successfully displacing their reference biological products. As I predicted last year, newer biosimilars are being adopted quickly, and their prices are declining rapidly.
Conveniently for HHS, ASPE's analysis stopped before the biosimilar boom began. The biosimilarmarket is finally beginning to fulfill its promise. The latest data show that provider-administered biosimilar drugs are successfully displacing their reference biological products. d/b/a Drug Channels Institute.
Under the heading “Facilitating Competition” are multiple initiatives designed to either hasten development of generic drugs or limit blockades to market. In the second category—limiting blockades to marketing—are efforts to limit blocking exclusivities and induced infringement liability.
Agreement includes commercializing biosimilar candidate of ophthalmology drug ranibizumab (Lucentis ® ) in Europe, Canada, Israel and global markets. Bioeq has in-licensed the exclusive global commercialization rights to FYB201 from the German biosimilar developer Formycon AG.
Food and Drug Administration approved the first interchangeable biosimilar insulin product, indicated to improve glycemic control in adults and pediatric patients with Type 1 diabetes mellitus and in adults with Type 2 diabetes mellitus. Semglee (insulin glargine-yfgn) is the first interchangeable biosimilar product approved in the U.S.
Sandoz, a Novartis division, today announced progress in the late-stage clinical development program for its proposed biosimilar aflibercept. The initiation of this study marks an important milestone in the development of our biosimilar aflibercept.
Gibbs — On March 21, 2024, the House Energy and Commerce held a subcommittee hearing titled “Evaluating Approaches to Diagnostic Test Regulation and the Impact of the FDA’s Proposed Rule.” By Ana Loloei & Jeffrey N. FDA, which was not invited to participate, would surely have concurred.
Sandoz strengthens pipeline expansion through partnership to develop and manufacture multiple biosimilars Sandoz, a global leader in off-patent (generic and biosimilar) medicines, today announced a multi-year partnership with Just – Evotec Biologics, the Seattle-based subsidiary of Evotec SE.
(HP&M), a leader in providing legal and regulatory support to the life sciences industries, today announced the appointment of Jeff Grizzel to the newly created position of Chief Marketing Officer (CMO). Grizzel holds a degree in Economics from High Point University and lives in Falls Church, VA.
With eight marketedbiosimilar medicines globally and 15+ molecules in pipeline, Sandoz is investing in future of biosimilars for patients and healthcare systems. The initiation of this study marks an important milestone in the development of our biosimilar aflibercept.
While that’s not to say that OTC hearing aids aren’t working to address the critical issue of hearing loss, it seems that almost two years is still not enough time to assess market impact, or to show that some of the promises of significant savings for large numbers of consumers have been realized.
Updated guidance on promotional labeling for biosimilars and interchangeables emphasizes a similar approach Today, the FDA issued a revised draft guidance on the development of promotional labeling for biosimilars, reference products, and—newly—interchangeable products. regarding its administration, preparation, storage, or safety).
What will the orphan drug market exclusivity haircut mean for industry? Just over a month ago, the European Commission released its proposal for the new pharmaceutical directive and regulation. provides a 10-year market exclusivity period. Orphan designation in the E.U.
Three of these articles focus on medical applications of Artificial Intelligence and Machine Learning (AI/ML) and explore FDA’s role in regulating such products. Gonzalez — The Wall Street Journal (WSJ) recently published a series of articles as part of its special report “What’s Ahead for Artificial Intelligence.”
However, ensuring that these structures comply with applicable tax laws and regulations is essential to avoid potential legal and reputational risks. Sophisticated valuation methodologies, such as the income, market, and cost approaches, are employed to estimate the fair value of individual patents or the entire IP portfolio.
With regulation and policy changes, specifically surrounding the DCSCA, IRA and serialization, the loss of exclusivity wave, adoption of low-WAC products affecting GTN and the rise of alternative distribution models, there has never been a more important time for industry to unite. Why should you attend this pivotal event?
By Riëtte van Laack — FDA regulates pet food similar to other animal foods. As anyone familiar with pet (and other animal) food regulation knows, many states require premarket label review and approval and registration of the manufacturer/distributor and/or product for a fee.
Lenz, Principal Medical Device Regulation Expert — For several years, FDA has requested that sponsors of drug or biologic led combination products identify essential performance requirements (EPRs) related to the device constituent in their applications. By Adrienne R. does not use this term.
Let’s just say, the smackdown—er, decision—eviscerates FDA’s approach to regulating flavored e-cigarettes. FDA also directed manufacturers to produce detailed marketing plans. Yet… Petitioners filed PMTAs for their flavored nicotine liquids, which included marketing and sales access restriction plans, with FDA before the deadline.
The announcement also states that FDA expects most future companion diagnostic and infectious disease IVDs would be regulated as class II devices, even if they are novel and require de novo classification. And as we note in that earlier blog , there is good reason to believe all of the above numbers are gross underestimates.
What we expect European regulators to do in May 2024 In this recurring feature, AgencyIQ, through public data and previous analysis, determines what European medicine and device regulators will likely do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods, and more.
Merck (NYSE: MRK), known as MSD outside the United States and Canada, announced today that Organon Finance 1 LLC plans to offer, subject to market conditions, euro-denominated senior secured notes due 2028, U.S. persons outside the United States in reliance on Regulation S under the Securities Act.
Baumhardt, Senior Medical Device Regulation Expert & Philip Won & Gail H. It states that FDA will review a PCCP for a ML-DSF as part of the initial marketing submission and “authorize” it in whole or in part, i.e., permit either all of the proposed modifications or only some of them. See 21 CFR 807.81(a)(3)
It merely says that “[t]he Hatch-Waxman Act and FDA regulations set forth the criteria for listing patents in the Orange Book” and that “Brand manufacturers are responsible for ensuring their patents are properly listed.” But it is not always clear which types of patents are eligible for listing in the Orange Book.
Baumhardt, Senior Medical Device Regulation Expert & Adrienne R. Lenz, Principal Medical Device Regulation Expert — FDA recently released a new eSTAR template for device pre-submissions and 513(g) Requests for Information, referred to as PreSTAR. PMA or 510(k)) is required. PMA or 510(k)) is required.
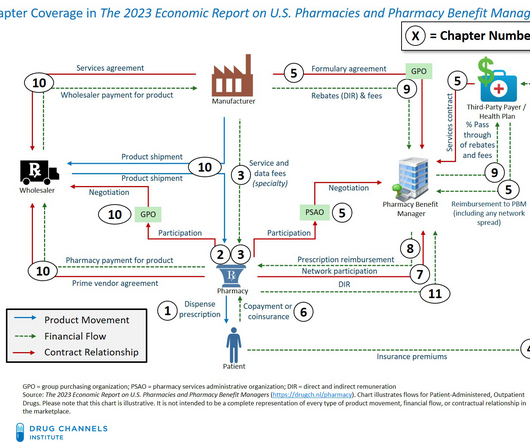
Pharmacies and Pharmacy Benefit Managers is a definitive, nonpartisan resource that includes the most current information about pharmacy dispensing channels, third-party payers, pharmacy benefit managers (PBMs), patients’ financial contributions, government regulations, and much more.
persons outside the United States in reliance on Regulation S under the Securities Act. In member states of the European Economic Area, this announcement is directed only at persons who are “qualified investors” within the meaning of the Prospectus Regulation.
As readers of our blog know, MoCRA was a significant change to regulation of cosmetics. Now in the second year of implementation, companies have started noticing the consequences as FDA implements the new requirements and develops regulations and guidance.
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
Our feed saw a number of rosy forecasts for mergers and acquisitions in FDA-regulated industries. In a “hot” deal market, its worthwhile reiterating the fundamentals of regulatory due diligence that don’t change, no matter how attractive the deal or how short the window is to seize the opportunity.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content