This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDA classifies it as a “nonsteroidal treatment” – not a gene therapy, but it affects gene expression. Results from the study that led to the FDAapproval appeared in The Lancet Neurology in April 2024 with commentary. A gene-based treatment would have to alter many cells to exert a noticeable effect.
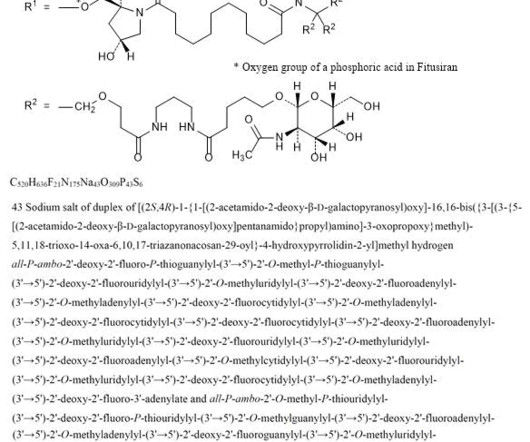
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 1] [2] Adverse effects The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal). [2] 26 March 2025.
FDAApproves Merck’s ENFLONSIA™ to Protect Infants from Severe RSV Illness Merck operating as MSD outside the United States and Canada, has received a significant regulatory milestone with the U.S. Food and Drug Administration (FDA) granting approval for ENFLONSIA™ (clesrovimab-cfor).
Dr Aaron Haubner, Senior Manager of North America Medical Affairs and Market Access at Terumo Blood and Cell Technologies , reveals that while promising new treatments emerge, urgent partnerships are needed to ensure this essential blood therapy reaches the patients who need it most.
Typical treatment has been chemotherapy and radiotherapy with an initially high overall response rate (ORR) but then rapid recurrence followed by poor prognosis. Toxicities were challenging enough to cause 13% of patients to reduce dose or skip doses and 3% to discontinue treatment. 4 patients (33%) had grade 4 adverse events.
Designed using the same fundamental mechanism as Biogen’s approved SMA treatment SPINRAZA (nusinersen), salanersen represents a next-generation approach aimed at greater potency and less frequent dosing—potentially requiring only once-yearly administration. Most adverse events (AEs) were mild to moderate in severity.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that the U.S.
FDAApproves Veklury (remdesivir) for the Treatment of COVID-19. Food and Drug Administration (FDA) has approved the antiviral drug Veklury (remdesivir) for the treatment of patients with COVID-19 requiring hospitalization. The speed and rigor with which Veklury has been developed and approved in the U.S.
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. NASDAQ: ATNX), a global biopharmaceutical company dedicated to the discovery, development, and commercialization of novel therapies for the treatment of cancer and related conditions, today announced that the U.S.
Xywav: A Low-Sodium Alternative with FDAApproval Xywav is a uniquely formulated, low-sodium oxybate therapy, and remains the only product of its kind approved by the U.S. It is also approved for adult patients with idiopathic hypersomnia (IH). mmHg (P=0.0003) In-office seated resting SBP decreased by −9.2
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
According to the data, the combination demonstrated a high ORR and a strong depth of response in a patient population with limited treatment options and poor outcomes under standard care. were penta-drug refractory, having been exposed to additional lines of treatment. Among these: 84.4% had previously received a bispecific antibody.
Novartis today announced the US Food and Drug Administration (FDA) has granted accelerated approval for Kymriah ® (tisagenlecleucel) for the treatment of adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two or more lines of systemic therapy.
FDAApproves Imcivree (setmelanotide) for Chronic Weight Management in Patients with Obesity Due to POMC, PCSK1 or LEPR Deficiency. Nasdaq:RYTM), a biopharmaceutical company aimed at developing and commercializing therapies for the treatment of rare genetic diseases of obesity, announced today that the U.S. BOSTON, Nov.
Despite its impact, there are currently no FDA-approved therapies for PMN , and treatment options are limited to non-specific and often toxic agents such as chemotherapy or general immunosuppressants. Despite the severity of PMN, nearly one-third of patients fail to achieve remission with current treatment strategies.
FDAapproves Pfizer’s LITFULO™ (ritlecitinib) for adults and adolescents with severe alopecia areata Pfizer Inc. Food and Drug Administration (FDA) has approved LITFULO™ (ritlecitinib), a once-daily oral treatment, for individuals 12 years of age and older with severe alopecia areata. with placebo.
Food and Drug Administration (FDA) has approved Repatha® (evolocumab) as an adjunct to diet and other LDL cholesterol (LDL-C)-lowering therapies for the treatment of pediatric patients aged 10 years and older with heterozygous hypercholesterolemia (HeFH) to scale back LDL-C. No new safety risks were identified.5
We are entering an area with significant unmet medical need since the current treatment paradigm for GBM remains bleak, as this aggressive and currently incurable form of brain cancer continues to claim high mortality rates. These statements relate to future events, future expectations, plans and prospects.
The Janssen Pharmaceutical Companies of Johnson & Johnson today announced preliminary data from the Phase 1 CHRYSALIS study evaluating RYBREVANT TM (amivantamab-vmjw) for the treatment of patients with non-small cell lung cancer (NSCLC) with mesenchymal-epithelial transition (MET) exon 14 skipping (METex14) mutations. 4] , [5] , [6].
We know from our research that the majority of men prefer an oral treatment to an injection,” Myovant CEO Lynn Seely said. The secondary endpoint for increasing lifespan for patients with metastatic prostate cancer came in at 74% still alive after treatment with Orgovyx compared to 75% of those on Lupron. .
Teva and MedinCell Announce FDAApproval of UZEDY™ (risperidone) Extended-Release Injectable Suspension, a Long-Acting Subcutaneous Atypical Antipsychotic Injection, for the Treatment of Schizophrenia in Adults Teva Pharmaceuticals, a U.S. The initiation of treatment requires no loading dose or oral supplementation.
Despite continuous innovations in the treatment landscape, unmet needs remain. If approved by the European Commission, ponesimod has the potential to help more people living with relapsing forms of MS.”. 5] The MAA will now be reviewed by the European Commission (EC) for the treatment of adults with RMS. vs. 10.4%).
a biopharmaceutical company engaged in the discovery and development of RNAi therapeutics against cancer and fibrotic diseases, today announced dose administration for the first patient in a Phase 2a clinical study of the company’s lead drug candidate, STP705, for the treatment of cutaneous basal cell carcinoma. GAITHERSBURG, Md.
ADUHELM is indicated for the treatment of Alzheimer’s disease. Treatment with ADUHELM should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. ADUHELM is indicated for the treatment of Alzheimer’s disease.
The approval is based on interim efficacy and safety data from the RAINBOWFISH study in newborns, which showed that the majority of pre-symptomatic babies treated with Evrysdi achieved key milestones such as sitting and standing with half walking after 12 months of treatment.
As defined by the National Cancer Institute, tumor board meetings convene multidisciplinary specialists to decide on the best treatment plan for new and complex cancer cases. FDA plans to issue a limited number of vouchers, without specifying an exact number as of yet. The vouchers will be issued to “companies aligned with U.S.
2] – The atogepant application demonstrates AbbVie’s longstanding commitment to providing multiple migraine treatment options, including BOTOX® (onabotulinumtoxinA), a preventive treatment for those with chronic migraine, and UBRELVY® (ubrogepant), an acute treatment for adults with migraine. .
Food and Drug Administration (FDA) has granted Orphan Drug Designation for HPN217 for the treatment of multiple myeloma. Harpoon has four drug product candidates in clinical development for the treatment of solid and hematologic malignancies based on its proprietary TriTAC platform. “I About the Phase 1/2 Trial for HPN217.
“The positive outcomes strengthen the efficacy and safety profile of erenumab as a migraine prevention treatment for patients with migraine.” “The positive outcomes strengthen the efficacy and safety profile of erenumab as a migraine prevention treatment for patients with migraine.” versus 38.9%). .
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of LN.
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. As a result, researchers now often use composite endpoints in lieu of the customary single primary endpoint.
FDA EMERGENCY USE AUTHORIZATION PRESCRIBING INFORMATION: Do not administer Pfizer-BioNTech COVID-19 Vaccine to individuals with known history of a severe allergic reaction (e.g., Vaccination providers must report Adverse Events in accordance with the Fact Sheet to VAERS at [link] by calling 1-800-822-7967.
Food and Drug Administration (FDA) has designated as a Fast Track development program the investigation of Brilacidin as a potential treatment for COVID-19. A molecular screening study of 11,552 compounds also supports Brilacidin as a promising novel coronavirus treatment. WAKEFIELD, Mass.,
Source link.
DEXTENZA is FDAapproved for the treatment of ocular inflammation and pain following ophthalmic surgery. The most common non-ocular adverse event was headache (1%). Ocular Therapeutix’s first commercial drug product, DEXTENZA, is FDA-approved for the treatment of ocular inflammation and pain following ophthalmic surgery.
Food and Drug Administration (FDA) blessing of VUITY ™ (pilocarpine HCl ophthalmic result)1.25 for the treatment of diplopia, generally known as age- related vague near vision, in grown-ups. There were no serious adverse events observed in actors entering VUITY in either the GEMINI 1 or GEMINI 2 study. adult population.
EST.
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of lupus nephritis (LN).
Food and Drug Administration (FDA) approved Actemra ® /RoActemra ® (tocilizumab) subcutaneous injection for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD), a debilitating condition with limited treatment options.
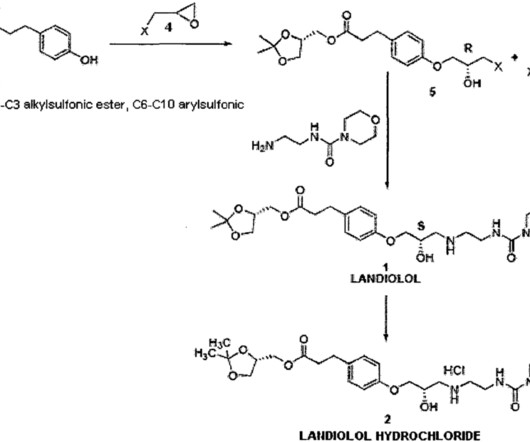
Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDAAPPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDAAPPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 9] It is used as landiolol hydrochloride.
an international research-focused healthcare Group (Chiesi Group), today announced final study results from the BRIDGE Phase III Open-Label, Switch-Over Clinical Trial Evaluating Pegunigalsidase Alfa for the Treatment of Fabry Disease. galactosidase-A product candidate under development for the treatment of Fabry disease. nM and 13.81
BYOOVIZ™ is the first FDAapproved ophthalmology biosimilar BYOOVIZ, priced 40% lower than LUCENTIS®, provides an equally effective and more affordable treatment option to patients suffering from retinal disorders BYOOVIZ will be commercially available through major distributors across the U.S. on July 1, 2022. Biogen Inc.
In this article, we review two case studies involving the successful use of RWD or RWE in advancing the clinical development of treatments for rare diseases. The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement.
. – If Authorized, Lenacapavir Would be the First Capsid Inhibitor and the Only HIV-1 Treatment Option Administered Twice Yearly –. Lenacapavir was generally well-tolerated, with no serious adverse events related to study drug and no study drug discontinuations through the 14-day period, including no discontinuations due to adverse events.
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. More targeted therapies may change which patients and cancers may benefit, since a given treatment may only impact very specific biomarkers and genetic profiles. This means study startup times can vary greatly.
Food and Drug Administration (“FDA”) for the treatment of agitation associated with delirium. Treatment choices are limited, and commonly used off-label therapies are not always effective or may result in prolonged, deep sedation. The Company plans to initiate a Phase 2 trial within the next several months. “We
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.







































Let's personalize your content