This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Clinical trials are expensive, slow and often limited by outdated design constraints. These digital twins are created for each trial participant using their baseline data – regardless of whether they are assigned to the placebo or treatment arm – and simulate how that individual would have responded under control conditions.
However, as we note in that post, the design, timing of initiation, and timely conduct of confirmatory trials are also important considerations in FDAs determination of whether accelerated approval is appropriate. This blog post focuses on interpreting these new authorities with respect to timely conduct of confirmatory trials.
Deep Dive Library Events Press Releases Topics Sign up Search Sign up Search Pharma Biotech FDA Clinical Trials Deals Drug Pricing Gene Therapy An article from Dive Brief Actithera draws new investors to radiopharma drug pitch The four-year-old biotech raised about $75 million in a Series A round that involved nine venture capital firms.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
Biogen Reports Promising Interim Phase 1 Results for Salanersen in Spinal Muscular Atrophy, Prepares for Registrational Trials Biogen has announced encouraging topline results from its Phase 1 clinical trial evaluating salanersen (BIIB115/ION306) , an investigational antisense oligonucleotide (ASO) therapy for spinal muscular atrophy (SMA).
A surrogate endpoint is a marker used in clinical trials as a substitute for a direct clinical outcome. Diagnostic biomarkers typically confirm or establish a diagnosis and are often used in selecting patient populations for clinical trials.
FDA Approves Streamlined Monitoring Requirements and REMS Program Removal for Bristol Myers Squibb’s CAR T Cell Therapies Breyanzi and Abecma, Marking Milestone Toward Expanding Access to Cancer Treatment In a significant regulatory development, Bristol Myers Squibb announced that the U.S.
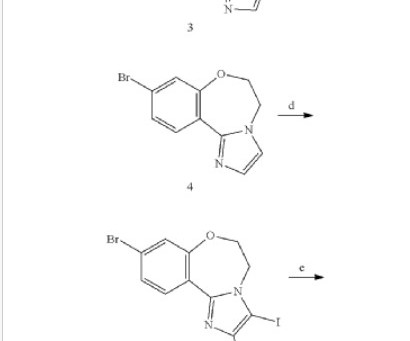
The presence of thienopyrimidine derivatives in several FDA-approved drugs and clinical trial candidates underscores their therapeutic potential and safety profile. The review elaborates on the primary approach for synthesis of thienopyrimidines, using thiophene derivatives or pyrimidine analogues.
FDA Approves Label Update for Lilly’s Amyvid, Expanding Its Role in Alzheimer’s Disease Diagnosis and Therapy Guidance In a major development for Alzheimer’s diagnostics, Eli Lilly and Company (NYSE: LLY) announced that the U.S. At the time, its use was primarily restricted to supporting the diagnostic process.
It is becoming increasingly evident that generative artificial intelligence (GenAI) is a resourceful tool for helping pharmaceutical companies reduce manual tasks required by clinical trials. Because LLMs are trained on extensive, internet-scale datasets, they can learn to identify contexts linking words and language.
Clinical Trials: Palazestrant is currently in clinical trials, including Phase 1/2 and Phase 3 studies, for the treatment of ER+, HER2- metastatic breast cancer. Promising Results: Preliminary results from clinical trials have shown promising antitumor efficacy and favorable pharmacokinetic properties for palazestrant.
The trial met both its primary and key secondary endpoints, signaling a potential breakthrough in a condition with few, if any, effective treatment options. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) —to expedite review and potential approval for this new indication.
Robust Clinical Trial Design : Clinical trials for biosimilars should be designed to demonstrate equivalence or non-inferiority to the reference product. This involves the use of statistical techniques such as equivalence and non-inferiority testing, as well as adaptive design approaches to reduce sample size and trial duration.
has announced encouraging interim findings from the pivotal Phase 3 VISIONARY trial evaluating sibeprenlimab in adults with Immunoglobulin A nephropathy (IgAN), a rare and progressive autoimmune kidney disorder. FDA Priority Review and Accelerated Development Pathway Sibeprenlimab has already drawn regulatory attention.
Clinical Trials: Phase 3 clinical trials have shown that nerandomilast can slow lung function decline in patients with IPF and PPF. Efficacy: The trials demonstrated that nerandomilast led to a smaller decline in forced vital capacity (FVC), a measure of lung function, compared to placebo.
The Untitled Letter notes that the confirmatory trial for LYTGOBI is currently ongoing and has not been completed; therefore, clinical benefit of LYTGOBI has not yet been confirmed. As a single-arm trial (i.e., The Untitled Letter states that a randomized controlled trial would be needed to assess delay in time to disease progression.
FDA Orphan Drug Designation for Antibody-Mediated Rejection in Solid Organ Transplantation The U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation (ODD) to riliprubart , an investigational immunology therapy developed by Sanofi, for the treatment of antibody-mediated rejection (AMR) in solid organ transplantation.
1] Medical uses In the US, sunvozertinib is indicated for the treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy. [1] 2 July 2025.
Food and Drug Administration (FDA) as a monotherapy for children with achondroplasia. The Week 26 data from the COACH Trial now show that TransCon hGH may further enhance these benefits. mg/kg/week) during the trial. Administered once weekly, TransCon CNP is currently under Priority Review by the U.S.
Ionis Begins Pivotal Phase 3 REVEAL Study of ION582 in Angelman Syndrome, Dosing First Patient in Global Trial Ionis Pharmaceuticals , Inc. This trial will build on previous positive data from the earlier Phase 1/2 HALOS study and is designed to rigorously assess the potential of ION582 as a disease-modifying therapy. “We
By organising and analysing this information, researchers can extract actionable insights that improve patient outcomes, data accuracy, drug efficacy and speed up trials. Clinical research generates vast amounts of diverse data from laboratory tests, patients, medical equipment, and outside sources.
Biogen Launches Global Phase 3 PROMINENT Trial Evaluating Felzartamab in Primary Membranous Nephropathy Biogen Inc. The trial is expected to complete in 2029. This randomized, open-label study will compare felzartamab with tacrolimus, a commonly used immunosuppressant, in a cohort of approximately 180 patients worldwide.
FDA Approves Merck’s ENFLONSIA™ to Protect Infants from Severe RSV Illness Merck operating as MSD outside the United States and Canada, has received a significant regulatory milestone with the U.S. Food and Drug Administration (FDA) granting approval for ENFLONSIA™ (clesrovimab-cfor). Key results from the CLEVER trial include: A 60.5%
The development of oncology drugs is a complex, multi-phase process, where safety, efficacy, and optimal dosing are determined progressively through clinical trials. To ascertain the RP2D effectively, Phase I trials are often split into two critical sub-phases, dose escalation and dose expansion.
Their unique suitability has made them valuable for evaluating pharmacokinetics, toxicology and safety in drug candidates before human clinical trials. NHP-C cardiomyocytes offer a more predictive and ethical platform, especially for drugs intended to advance from animal testing to first-in-human trials.
Xywav: A Low-Sodium Alternative with FDA Approval Xywav is a uniquely formulated, low-sodium oxybate therapy, and remains the only product of its kind approved by the U.S. Food and Drug Administration (FDA) for the treatment of cataplexy or excessive daytime sleepiness (EDS) in patients aged seven years and older with narcolepsy.
FDA Approves ANDEMBRY (garadacimab-gxii) The First Once-Monthly Prophylactic HAE Therapy Targeting Factor XIIa CSL a leading biotechnology company with a strong track record of developing innovative medicines for patients with rare and serious disorders, today announced that the U.S.
4] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [7] Jump up to: a b c d e f g h i j k l “FDA approves treatment of amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene” (Press release). Food and Drug Administration (FDA). 3 November 2006. 3 June 2024.
This was in 2006, at a time when the FDA guidances on these topics had not yet been published. Over time, this group of professionals evolved and grew, having regular stakeholder interactions with the FDA and Controlled Substances staff to discuss requirements for drug developers. corticosteroids, beta-blockers, antidepressants).
Though it may not be used across a broader patient population, results from Arvinas and Pfizer’s trial of vepdegestrant offer some validation for degraders’ potential. The two partners, despite filing for approval, have pared back two other planned trials of vepdegestrant and Arvinas cut one-third of its staff in May.
Safety Profile Consistent with Previous Studies : The safety profile of rilzabrutinib was well-understood from previous trials, with no new safety signals. Fast Track and Orphan Drug Designations from FDA The U.S.
With global regulators including the FDA, EMA, and MHRA signaling alignment and implementation in 2025 [FDA, 2025; EMA, 2025; MHRA, 2025], sponsors must act now to align their strategies with the new expectations. FDA Statement on Adoption of ICH E6(R3). What needs to change? Learn more References: ICH Harmonised Guideline.
The technology has already been successfully tested in a clinical trial for another rare genetic disease. The scientists delivered the prime editors to cells in mice using clinically validated viruses called AAVs, which are already used in FDA-approved gene therapies targeting brain cells. It was incredibly exciting to see that data.”
We are encouraged by the clinical activity observed in early-phase trials, including uveal melanoma, a rare eye cancer with limited treatment options. Additionally, we are preparing to launch several other trials over the coming months. These trials offer significant potential to help even more patients.
19] Society and culture Legal status In October 2024, the US Food and Drug Administration (FDA) approved inavolisib for the treatment of PIK3CA -mutant breast cancer based on the results from the INAVO120 trial. [3] 3] [6] [20] [21] The drug application was granted priority review and breakthrough therapy designations by the FDA. [3]
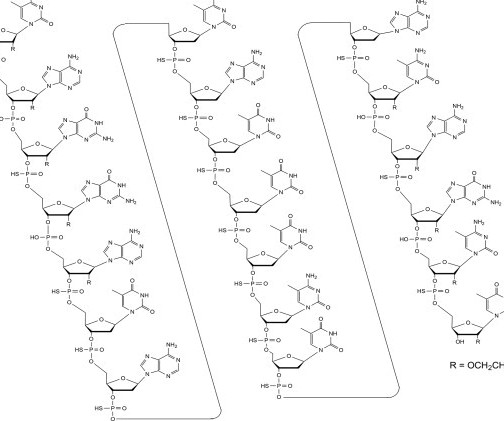
Food and Drug Administration (FDA) issued two guidance documents outlining the necessary evaluations during the clinical development of oligonucleotide therapeutics: Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics and Nonclinical Safety Assessment of Oligonucleotide-Based Therapeutics .
4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness. [5] 4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness. [5]
Two large and randomized clinical trials, IMpower-133 and CASPIAN, showing statistically significant improvements in outcomes when chemotherapy was combined with atezolizumab or durvalumab, respectively, in the first-line treatment of ES-SCLC. This Phase 3 trial compared tarlatamab with chemotherapy as second-line treatment.
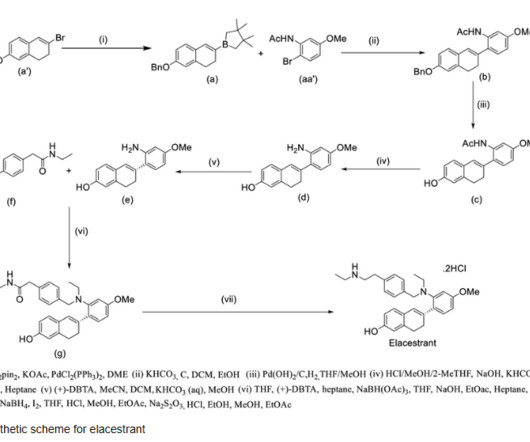
1] History The efficacy of elacestrant was evaluated in the EMERALD trial, which was a randomized, open-label, active-controlled, multicenter study involving 478 postmenopausal women and men with ER-positive, HER2-negative advanced or metastatic breast cancer. Food and Drug Administration (FDA). 1] [4] It is taken by mouth. [1]
2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] The FDA has long supported development of non-opioid pain treatment. acting director of the FDA’s Center for Drug Evaluation and Research.
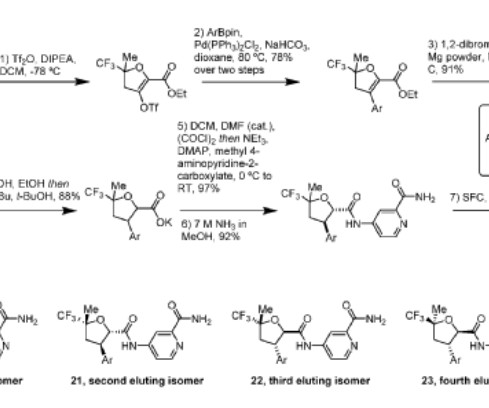
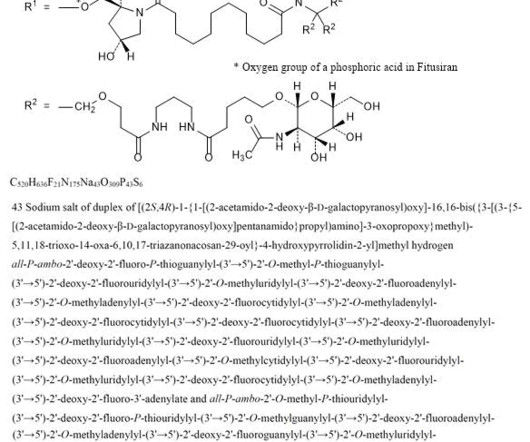
2] History The efficacy and safety of fitusiran were assessed in two multicenter, randomized clinical trials which enrolled a total of 177 adult and pediatric male participants with either hemophilia A or hemophilia B. [2] The FDA granted the approval of Qfitlia to Sanofi. Fitusiran 1711.0g/mol, Fitusiran 1711.0g/mol, 26 March 2025.
No B7-H3-directed therapeutics have received FDA or other approvals yet but the wave is building, especially with ADCs. That trial is in second-line NSCLC. The sheer number of programs creates a complex and competitive landscape on top of the diverse clinical applications being tested.
In an era where clinical trials are increasingly global, it’s more imperative than ever to leverage international expertise. Data and safety monitoring boards (DSMBs), also known as data monitoring committees (DMCs), play a critical role in overseeing a clinical trial’s safety and efficacy. local standards of care).
Together, these technologies have entered at least 19 clinical trials, with clinical results from seven of those trials reported so far — all showing that the base edit or prime edit resulted in patient benefit. Of the 19 base editing and prime editing clinical trials underway now, more are taking place outside the U.S.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content