This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
FDAApproves Streamlined Monitoring Requirements and REMS Program Removal for Bristol Myers Squibb’s CAR T Cell Therapies Breyanzi and Abecma, Marking Milestone Toward Expanding Access to Cancer Treatment In a significant regulatory development, Bristol Myers Squibb announced that the U.S.
FDAApproves Tablet Formulation of BeOne’s BRUKINSA® for All Approved Indications, Offering Greater Convenience for Patients with B-cell Cancers BeOne Medicines Ltd. Food and Drug Administration (FDA). a global oncology-focused biopharmaceutical company, has received a significant regulatory milestone from the U.S.
FDAApproves Merck’s ENFLONSIA™ to Protect Infants from Severe RSV Illness Merck operating as MSD outside the United States and Canada, has received a significant regulatory milestone with the U.S. Food and Drug Administration (FDA) granting approval for ENFLONSIA™ (clesrovimab-cfor).
Currently, more than 20% of FDA-approved monoclonal antibodies (mAbs) fall into this category, reflecting a growing industry trend. Despite these benefits, the development and commercialization of high-concentration biologics come with formidable challenges. WuXiHigh™ 2.0: Pushing the Boundaries With WuXiHigh™ 2.0, Source link
This decision marks the 10th productapproval supported by AGC Biologics’ Milan site—a Cell and Gene Center of Excellence that has now achieved regulatory success with both the European Medicines Agency (EMA) and the U.S. FDAapproval in November 2024, and a U.K.
Xywav: A Low-Sodium Alternative with FDAApproval Xywav is a uniquely formulated, low-sodium oxybate therapy, and remains the only product of its kind approved by the U.S. It is also approved for adult patients with idiopathic hypersomnia (IH).
FDAApproves Label Update for Lilly’s Amyvid, Expanding Its Role in Alzheimer’s Disease Diagnosis and Therapy Guidance In a major development for Alzheimer’s diagnostics, Eli Lilly and Company (NYSE: LLY) announced that the U.S.
The goal is to boost SMN protein production more effectively and offer a more convenient, once-a-year dosing schedule , thereby improving long-term disease management and patient compliance. Some patients demonstrate suboptimal clinical outcomes or experience treatment fatigue due to ongoing, frequent dosing regimens.
3] Tofersen is an antisense oligonucleotide that targets the production of superoxide dismutase 1 , an enzyme whose mutant form is commonly associated with amyotrophic lateral sclerosis. 3] Tofersen was approved for medical use in the United States in April 2023, [3] [6] and in the European Union in May 2024. [4] 25 April 2023.
Delays in Abbreviated New Drug Application (ANDA) submissions or setbacks during FDA review can mean missed market opportunities, especially in competitive therapeutic areas.
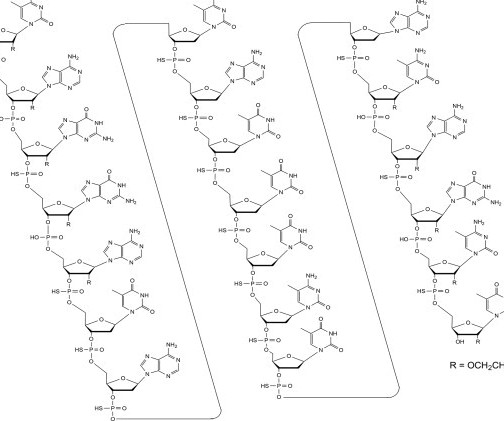
Finally, the acetate group is cleaved under basic conditions to give crude Vamorolone, which was recrystallized from iPrOH to obtain the pure product. The product was filtered off, washed with H 2 O (3 x 0.5 The product was filtered off, washed with H 2 O/MeCN 4:1 (2 x 0.5 and DMAP (16.1 g, 0.132 mmol, 0.10 HCI (950 mL, 0.95
The announcement is particularly relevant given the lack of FDA-approved treatment options for AMR, a complication that poses a critical threat to transplant success and long-term graft survival. This form of rejection can occur early or late after transplantation and is notoriously difficult to treat.
However, in 2012 it once again failed to convince the FDA of its qualities for treating anxiety and depression. [5] 5] In December 2015, the FDA once again gave gepirone a negative review for depression due to concerns of efficacy. [12] 12] However, in March 2016, the FDA reversed its decision and gave gepirone ER a positive review. [13]
There are over 7,000 rare diseases affecting more than 30 million people in the United States, and despite the FDA'sapproval of over 600 treatments for rare diseases since signing the Orphan Drug Act into law in 1983, most rare diseases still do not have a treatment.
The next step was to adapt cell culture techniques to enable the production of effective CAR T cells for infusion to patients. In 2012, the University of Pennsylvania team and Novartis launched a collaboration to further develop the technology, which led to the FDAapproval for the first commercial CAR T-cell therapy in 2017.
3] Inavolisib was approved for medical use in the United States in October 2024. [3] The residue was purified via flash chromatography on silica gel (eluted: 15% ethyl acetate in petroleum ether) to give crude product which was washed with 2.4 g (90%) of the crude title product as a colorless oil. Hz, 1H), 3.71-3.57
This mechanism has the potential to partially restore UBE3A protein production in neurons, addressing the root cause of the disorder. ION582 works by inhibiting a natural antisense transcript that suppresses the expression of the paternal UBE3A allele, thereby allowing it to be reactivated.
As clinical adoption accelerates and regulatory approvals increase, there are now four FDA-approved T cell engagers and 17 bispecific antibody therapies available on the global market. With regulators focused on product consistency, these capabilities are essential for achieving clinical milestones and commercial viability.
Despite its impact, there are currently no FDA-approved therapies for PMN , and treatment options are limited to non-specific and often toxic agents such as chemotherapy or general immunosuppressants. Felzartamab is currently an investigational product and has not yet received approval from any regulatory authority.
Casgevy , the first FDA-approved CRISPR-based therapy, is also an ex vivo gene therapy; it aims to cure sickle cell disease by editing a patient’s blood-producing stem cells so they generate functional fetal hemoglobin, then returning these corrected cells to the bloodstream. Clearly, this vector works.
The FDAs Oncology Center of Excellence (OCE) plays a pivotal role in fostering innovation, collaboration, and efficiency in the development and evaluation of oncology products. Determining a new products pathway The OCE does not receive regulatory applications directly from sponsors who are developing new cancer therapies.
5] History Pirtobrutinib is manufactured by Eli Lilly and Company and was approved by the US Food and Drug Administration in January 2023, for the treatment of mantle cell lymphoma that has become refractory to other BTK inhibitors. [13] 14] The applicant for this medicinal product is Eli Lilly Nederland B.V. [14] 27 January 2023. .
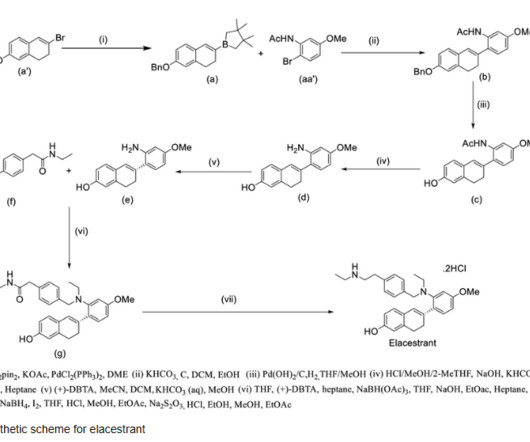
2] The FDA granted the application for elacestrant priority review and fast track designations. [2] Jump up to: a b c d e f g “FDAapproves elacestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer” U.S. Food and Drug Administration (FDA). 8 February 2023. 27 January 2023.
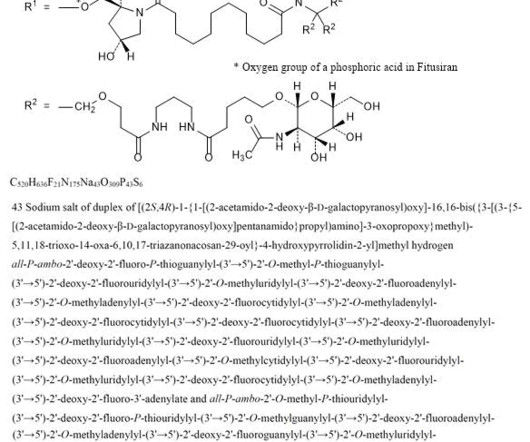
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 2] The fixed dose of fitusiran is not approved because it led to excessive clotting in some participants. [2] 2] The US Food and Drug Administration (FDA) granted the application for fitusiran orphan drug and fast track designations. 26 March 2025.
Currently, three FDA-approved disease-modifying drug therapies are available: hydroxyurea, crizanlizumab and L-glutamine, though each has limitations that affect patient compliance. The use of FDA-approved medications for preventing vaso-occlusive events in sickle cell disease. JAMA Netw Open. 2023 Nov 1;6(11):e2344546.
Her deep experience in managing government contracts, navigating regulatory pathways, and driving product development to FDAapproval makes her an ideal leader. Kenton Gregory, Chairman of Synedgen’s Board of Directors. I look forward to working closely with Dr.
1] [2] It was developed by Vertex Pharmaceuticals , [5] and was approved for medical use in the United States in January 2025. [2] 2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] Food and Drug Administration (FDA).
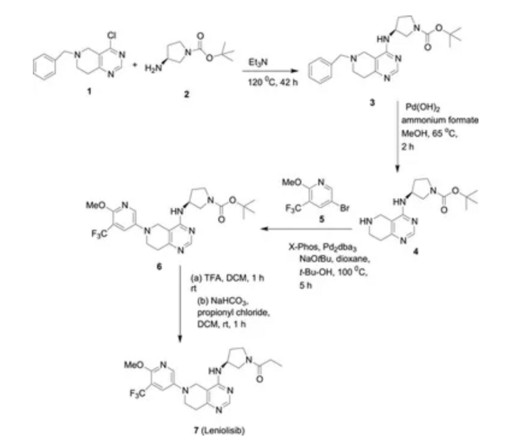
5] Leniolisib was approved for medical use in the United States in March 2023. [5] 5] [7] [8] It is the first approved medication for the treatment of activated PI3K delta syndrome. [5] 5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [9] “Leniolisib: First Approval” Drugs.
The obtained crude product was purified by silica gel (60-120 mesh) column chromatography to get repotrectinib asawhite solid (0.18 To a solution of the acid product in DCM (4 mL) was added 4 M HCl in 1,4-dioxane (2.0 To a solution of the de-Boc product and FDPP (27.62 was granted FDAapproval on November 15, 2023.
2] Crinecerfont was approved for medical use in the United States in December 2024. [2] 2] [3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [4] 2] The FDA granted the approval of Crenessity to Neurocrine Biosciences, Inc. [2] Food and Drug Administration (FDA) (Press release).
WINREVAIR is the first FDA-approved activin signaling inhibitor for PAH. The product was developed under a licensing agreement with Bristol Myers Squibb. The drug works by rebalancing signaling pathways that regulate vascular remodeling, with the goal of reducing pro-proliferative responses in pulmonary blood vessels.
The crude product was purified by flash chromatography (120 g column, 0–100% (10% ethanol in ethyl acetate) in isohexanes to afford two regioisomers: 21a (2.03 3] Society and culture Legal status Sebetralstat was approved for medical use in the United States in July 2025. [1] mmol, 47% yield) as an off-white solid and 21b (350 mg, 0.92
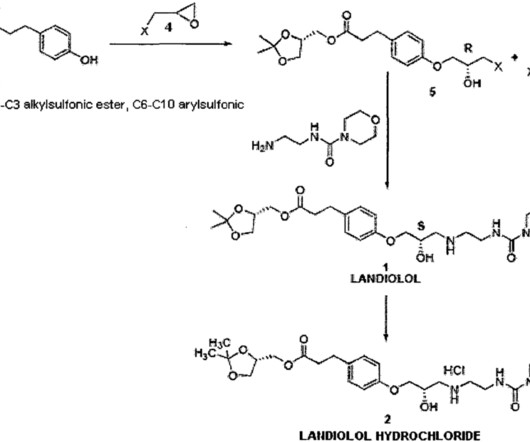
Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDAAPPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 After addition of the acid, the solvent is evaporated off and the product is crystallized by adding 1 20 volumes of a polar solvent, preferably acetone. 20 November 2023. 21 December 2022.
Today, a single injection of an FDA-approved gene therapy, called Hemgenix , cures this disease. Teitelman continues: A company that has mortgaged its future or lacks development skills or has no money left or has a product no one wants faces a limited future, no matter how powerful its technology.
The suspension is stirred and filtered; the product cake is washed with purified water and dried at room temperature in a nitrogen stream and then under vacuum. The product is isolated by filtration and washed with purified water. The product is isolated by filtration and washed with purified water. Crute, J.J.; Tsurumi, T.;
.— Jonathan Gardner The Food and Drug Administration on Monday approved broader use of Apellis Pharmaceuticals ’ rare disease drug Empaveli, clearing it for the kidney diseases C3 glomerulopathy or primary immune complex membranoproliferative glomerulonephritis in people 12 years of age or older.
Further, the method of Non-Patent Document 2 has a problem that the reaction time is long and many by-products such as biopterin in which BH4 is oxidized and deoxysepiapterin from which the β-position hydroxyl group of the side chain is eliminated are also generated. The crude product was purified by flash chromatography to give 2.09
The reaction mixture was concentrated under reduced pressure, and the crude product was redissolved in dichloromethane and washed with aqueous NaHCO 3. The organic layer was dried over Na 2 SO 4 and concentrated, and the resulting crude product which was used directly in the next step (767 mg, crude). mL, 16 mmol). mL, 16 mmol).
Verify the manufacturer's reputation : Research the company behind the generic medication to ensure they have a track record of producing high-quality products. Check the FDAapproval status : Make sure the generic medication has been approved by the FDA and meets all necessary safety and efficacy standards.
Generic drugs are pharmaceutical products that contain the same active ingredients as their brand-name counterparts. Explain the FDAApproval Process Many patients are unaware of the rigorous approval process generic drugs must undergo. What Are Generic Drugs?
A recent analysis from NORD showed that among the 39 rare pediatric diseases for which vouchers were awarded, only three had any FDA-approvedproducts on the market before the program’s enactment.
Lilly was the first company to introduce a self-pay solution for an FDA-approved obesity medication, and we remain passionate about expanding coverage for Zepbound. Zepbound contains tirzepatide and should not be used in combination with other tirzepatide products or with other GLP-1 receptor agonists.
Livornese — After teasing a new rapid review pilot program for the past few weeks, on June 17, 2025, FDA officially announced the Commissioner’s National Priority Voucher (“CNPV”) program to expedite new drug and biologic (but not device or drug-device combination product) reviews. months, ranging from 0.4 months to 5.9.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






































Let's personalize your content