This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Farquhar This is the first in a series of blog posts on tips for successfully handling an FDA inspection. Using publicly available examples, these lessons will illustrate potential pitfalls and strategies for interacting with FDA during and after an inspection. FD&C Act 501(j).
But growing ethical scrutiny, supply shortages, cost burdens, scientific innovation and regulatory shifts like the US Food and Drug Administration (FDA)’s new alternative methods roadmap are bringing the continued reliance on live NHPs into question, and opening the door to next-generation solutions that could eventually replace them altogether.
This was in 2006, at a time when the FDA guidances on these topics had not yet been published. Over time, this group of professionals evolved and grew, having regular stakeholder interactions with the FDA and Controlled Substances staff to discuss requirements for drug developers. corticosteroids, beta-blockers, antidepressants).
Vamorolone was approved by the FDA in October 2023 for the management of DMD in patients ≥2 years of age. 2017, 82, 11961−11980. (77) 2017, 82,11961−11980. 19] The US Food and Drug Administration (FDA) approved vamorolone based on evidence from a single clinical trial of 121 boys with DMD who were 4 to <7 years of age.
As the CEO of iOnctura, an innovative oncology biopharmaceutical company she co-founded in 2017, Catherine has played a key role in advancing the development of highly targeted small molecules aimed at revolutionising cancer treatment. So, with a huge amount of courage and commitment, I co-founded iOnctura in 2017.
4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness. [5] 4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness. [5]
A similar CAR from another company, Kite Pharmaceuticals (now a Gilead company), was approved by the FDA in 2017 as the second commercial CAR T-cell therapy. After their clinical results, their technology was licensed to Juno Therapeutics (later acquired by Bristol Myers Squibb.)
Compounds useful as kinase inhibitors (WO 2017/103611 A1). 1] [11] In December 2023, the US Food and Drug Administration (FDA) expanded the indication for pirtobrutinib to include the treatment of adults with chronic lymphocytic leukemia or small lymphocytic leukemia. [7] Food and Drug Administration (FDA). 2] PATENTS Guisot, N.
The FDAs Oncology Center of Excellence (OCE) plays a pivotal role in fostering innovation, collaboration, and efficiency in the development and evaluation of oncology products. Provides the Oncology Dosing Toolkit. Resulting ideas will be shared with those interested in implementing them.
5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [9] 5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [9] World Health Organization (2017). Food and Drug Administration (FDA) (Press release). mL) and the mixture stirred at rt for 1 h.
The treatment, now known as Casgevy, became the first CRISPR-based therapy to gain FDA approval, in 2023. Notably, this has led to the development of new medicines to treat genetic diseases — Casgevy was the first of these to gain FDA approval and is used to treat two blood disorders, called sickle cell disease and beta thalassemia.
In 2017, small and medium-sized biotech companies accounted for 51% of FDA market approvals, while large pharma companies were the originators in only 28% of approvals[4]. The pharmaceutical landscape is evolving rapidly, with small and medium-sized biotech companies increasingly taking center stage in innovation.
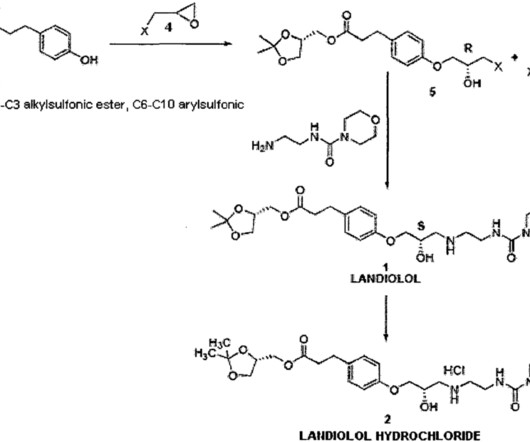
Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDA APPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 FDA Approves AOP Health’s Rapiblyk (landiolol) for Atrial Fibrillation and Atrial Flutter in the Critical Care Setting” (Press release). Food and Drug Administration (FDA).
US FDA granted fast track designation for pritelivir in 2017 and breakthrough therapy designation 2020. 7] If the virus also acquires resistance to foscarnet, then there is currently no FDA approved treatment. This is particularly important in immune compromised patients. PAPER By: Carta, Fabrizio ; et al.
Karst The FDA Reduction-in-Force (Termination)or RIF(T)announced last week has resulted in countless stories in the press and on personal LinkedIn accounts from those RIFd. As folks steeped in the world of generic drugs And Hatch-Waxman know, theres a lot that happens before FDA can take action on an ANDA.
According to the FDA, these REMS requirements are no longer necessary to ensure safe use of these CAR-T therapies as both physicians and hospitals are now well versed in managing the two syndromes most commonly associated with the drugs. Reporting rates for CRS and ICANS have “remained stable,” the FDA added.
Baumhardt, Principal Medical Device Regulatory Expert In January 2025, FDA posted the 2024 Annual Report concerning the Accreditation Scheme for Conformity Assessment (ASCA) program as required by Medical Device User Fee Amendments of 2017 (MDUFA IV).
One expert views Amtagvi’s approval as a catalyst for further investment in TIL therapies, akin to how Kymriah’s 2017 clearance buoyed CAR-T treatment.
Every six months, we do a look-back to see what has changed in the way that FDA is communicating. The number of press releases issued by FDA during the first six months of the year declined to 101 from the 125 issued by June 30 of last year, the lowest number at mid-year since 2017. And there is more to tell.
Periodically I write a posting to look back at what FDA is talking about to get some perspective. So let’s take a look: Volume – Let’s look first at the numbers of releases issued by FDA for the first six months of the year. Content – Next up – what did FDA talk about?
Gibbs & Ana Loloei & Véronique Li, Senior Medical Device Regulation Expert — FDA has long touted the use of real-world evidence ( RWE ). FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices.
BY LAURA DIANGELO, MPH Under the Federal Food, Drug and Cosmetic Act, facilities may not deny, delay or limit FDA inspections of facilities, or else the products they make may be considered adulterated. The FDA has now finalized a revision to its guidance on the subject to provide more examples and incorporate device considerations.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
Schwartz — FDA recently published a Federal Register (FR) Notice [ Docket No. FDA-2024-N-3945 ] announcing the publication of a draft strategy document, for public comment, outlining specific actions FDA plans to take to facilitate the use of innovative manufacturing technologies.
FDA unveils long-awaited Patient Medication Information proposed rule Since 2017, the FDA has been working on a proposal to create a new type of patient-focused labeling for certain outpatient drug products that would be specifically targeted for patient use. Fill out the form to read the full article.
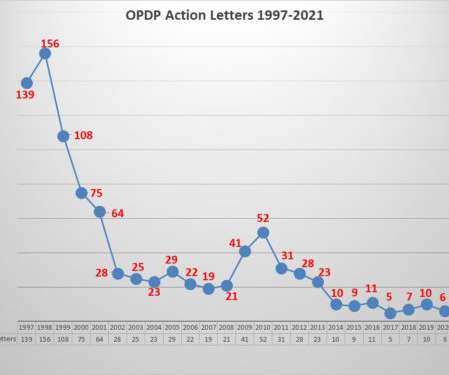
Once again it was a quiet year with respect to the FDA’s Office of Prescription Drug Programs (OPDP). The only other time in the past 24 years when enforcement was this low was in 2017. FDA has traditionally taken the view that if it is wrong on television DTC, then it is wrong on TikTok. in November 2009.
Photo by the CDC/FDA. The FDA has approved a new indication of Trelegy Ellipta for the treatment of asthma and patients with chronic obstructive pulmonary disease. . GSK submitted the application to the FDA back in October, and offered data from its Phase 3 CAPTAIN study to prove its effectiveness.
Richardson — While Covid is in the rear view for most of us, FDA has had a tough time shaking off the effects of the pandemic on its inspection output. In March 2020 , FDA temporarily postponed all foreign and domestic and routine surveillance facility inspections. Inspections went down—way down—during the pandemic.
Tobolowsky — Much has changed since the long-gone days of 2017. The Washington Nationals won the World Series, Presidential administrations have come and gone, and FDA has added new meeting types and formats to its menu. And so, FDA has issued a new draft guidance to bring everyone up to speed on formal meetings under PDUFA.
Through its Human Drugs Advisory Committee process FDA has a vast array of outside experts to consult on matters related to product approval as well as questions about policy or safety issues concerning approved products. FDA Advisory Committees (AdComms) ain’t what they used to be. In-person FDA AdComms were not really fun.
Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Javitt & Michael D. Shumsky & Philip Won & Adrienne R. Gaulkin & Jeffrey N.
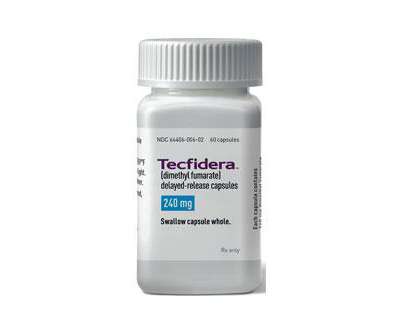
The FDA has approved its first generic of Biogen’s multiple sclerosis (MS) treatment Tecfidera, awarding authorisation to Mylan which is launching the drug in a dimethyl fumarate delayed-release oral solid formulation, both in 120mg and 240mg doses.
“It’s important that Impulse Dynamics acted quickly in a joint effort with the FDA to remove this potential barrier standing in the way of critical diagnostic procedures for a subset of patients with HF. Impulse Dynamics, based in Marlton, N.J.,
After suing each other in patent litigation and Avadel’s suit against FDA challenging the Agency’s authority to compel patent certifications, it’s Jazz’s turn to sue FDA. Approximately six weeks after FDA approved Lumryz and issued its clinical superiority decision , Jazz filed a Complaint against FDA in the District Court of D.C.
Metabolism of 2023 FDA Approved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. are major metabolites according the FDA Metabolites in Safety Testing guidelines). 2] Iversen et al.,
FDA finalizes guidance extending inspection authority to medical devices Under the Federal Food, Drug and Cosmetic Act, facilities may not deny, delay or limit FDA inspections of facilities, or else the products they make may be considered adulterated. Section 707 of FDASIA – now contained in the U.S.
Food and Drug Administration (FDA) has granted Fast Track Designation (FTD) for efanesoctocog alfa, previously known as BIVV001 (rFVIIIFc-VWF-XTEN), in patients with hemophilia A. Efanesoctocog alfa was granted orphan drug designation by the FDA in August 2017 and the European Commission in June 2019.
Drug manufacturers have had electronic systems in place since 2017. However, FDA recognizes that some “technical and operational issues,” including those involving downstream trading partners and other “affected stakeholders,” may not be fully resolved by the deadline. So what is FDA going to do? By Karla L. Guidance at 4.
Our approach to seizure liability screening is especially pertinent in the context of the recent FDA modernization act which allows applicants to use methods other than animal testing to establish drug safety and effectiveness. 2017 Jan;155(1):234–47. 2017 Jul 6;21(1):14–7. 2017 Feb;16(2):115–30. Toxicol Sci. Rockley KL.
The FDA rejected a psychedelic sponsor’s bid for approval. Here are six industry takeaways On August 9, Lykos Therapeutics received a Complete Response Letter (CRL) from FDA stating the agency will not currently approve its application for midomafetamine (MDMA) to treat post-traumatic stress disorder.
For first time, FDA releases OTC drug user fees prior to start of government fiscal year The FDA today unexpectedly unveiled certain user fees under its OTC Monograph User Fee Program (OMUFA), its nonprescription drug funding and performance mechanism. Without sufficient resources, the FDA is unable to do so.
Food and Drug Administration (FDA) has approved FoundationOne®Liquid CDx, Foundation Medicine’s comprehensive pan-tumour liquid biopsy test for patients with solid tumours. The test is FDA-approved to report short variants in 311 genes including rearrangements and copy number losses in BRCA1 and BRCA2 genes.
On the premarket side, in addition to the preparation of pre-submission and submission materials for FDA, Jennifer assists clients in responding to inquiries from FDA during the review process and participating in meetings with clients to address FDA concerns. Ellison, Managing Director at HPM.
Baumhardt, Senior Medical Device Regulation Expert & Philip Won — The Center for Devices and Radiological Health’s (CDRH) Standards and Conformity Assessment Program (S-CAP) encourages medical device sponsors to use FDA-recognized voluntary consensus standards in their product submissions.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content