This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
On July 21, the European Commission (EC) officially granted marketing authorization for AUCATZYL® (obecabtagene autoleucel, or obe-cel), a CAR-T cell therapy developed by Autolus Therapeutics. Food and Drug Administration (FDA).
This ability to activate pathways could be possible for almost all proteins, however the biggest differentiation was to consider areas that were hard to drug – transcription factors, ion channels, intracellular proteins, etc. Is it more important to have a clinical meaningful drug candidate OR convenience?
Vor Biopharma licensed a drug in June 2025 that targets proteins essential to B cell survival. Love Employee via Getty Images Dive Brief: Vor Biopharma is licensing rights to an immune disease drug from Chinese biotechnology company RemeGen, it said Wednesday, a little over one month after announcing plans to review strategic alternatives.
Fundamentally, even during tighter markets like today, great talent is always a very constrained resource, and spreading it too thinly across too many companies isn’t good for the ecosystem. Further, the competitive intensity in certain areas makes the “clinical do-ability” too challenging, even if those drugs could likely be beneficial.
In the ever-evolving field of medical science, artificial intelligence (AI) has emerged as a revolutionary tool, particularly in drug discovery. As a newer alternative to the time-consuming nature of traditional candidate screening and drug discovery, AI-designed drugs have started to make their mark, promising quick and efficient results.
By Faraz Siddiqui — Last Friday, the Delaware District Court rejected AstraZeneca’s lawsuit against the Medicare Drug Price Negotiation Program enacted under the Inflation Reduction Act (IRA) and CMS’s guidance implementing it. Opinion at 17. contradicts the plain text of the statute and therefore must be set aside.”
Food and Drug Administration’s Office of Orphan Products Development has granted Orphan Drug Designation to Uttroside-B , a small molecule chemotherapeutic for the treatment of hepatocellular carcinoma (HCC), the most common form of liver cancer. billion by 2027.
NEW YORK , Jan.
Q BioMed Inc.
These products are devices as defined in section 201(h) (1) of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of the Public Health Service Act, including when the manufacturer of these products is a laboratory.
This database was created to share post-market surveillance information across E.U. Ultimately, this module will interact with CTIS , the drug clinical trial information system. The independent auditing schedule showed publication of full functionality in mid-2027. countries and Notified Bodies (NBs).
Contract research organizations (CROs) are an integral partner of the drug development process, as they play a pivotal role supporting clinical trial conduct for pharmaceutical, biotechnology, and medical device sponsor companies. billion, projected to grow at a compound annual growth rate (CAGR) of 6.9% between 2023 and 2032.
Class A devices needed to be IVDR-ready on the original date (May 26, 2022), but class D had until May 2025, class C until May 2026, and class B and sterile class A until May 2027. In-house tests (in the U.S. called laboratory-developed tests manufactured and used in a single lab) had until May 26, 2024.
This activity could increase (or reduce the size of any decrease in) the market price of the Company’s common stock, the notes or the Company’s 2.50% Convertible Senior Notes due 2027 at that time. per share on the Nasdaq Global Select Market on January 25, 2021.
How have pre-submission meetings for generic drug applicants changed under GDUFA III? Under GDUFA III, the scope and purpose of pre-submission meetings for generic drug developers has changed. One of the core goals of the GDUFA program has been to increase the efficiency of the generic drug review program.
This means that the medical device frameworks – including the pre-market pathways and evidentiary requirements, as well as post-market quality system frameworks and reporting requirements – are applied to the vast majority of all (CDRH-regulated) IVD products. Are there implications for drug or therapeutic developers?
Food and Drug Administration (FDA) made public a potentially game-changing proposal concerning the regulatory framework for laboratory-developed tests (LDTs). years after publication; no earlier than October 1, 2027) End of Enforcement Discretion for: Premarket review for high-risk IVDs. On September 29, 2023, the U.S. Phase 4 (3.5
The agency further proposed a five “stage” phaseout of its current enforcement discretion, which would walk in the medical device regulatory requirements – from adverse events reporting to pre-market review – over four years. Under the proposed rule, these tests would be able to continue operating under the current status quo.
The registration database, EUDAMED , isn’t yet complete d, with just half of its planned modules released to production – actor registration, UDI and device registration, and Notified Bodies and certificates are live; still being worked on are the modules for clinical and performance studies, vigilance, and market surveillance.
In 2003, the CTD became the mandatory format for new drug applications in the EU and Japan, and the “strongly recommended format” for submission of applications to the FDA. The format and organization of the CTD is outlined in the ICH M4 Guideline.
After the fierce competition between Humira (adalimumab) biosimilars, manufacturers are keeping a close eye on adoption dynamics for biosimilars of Johnson & Johnson’s autoimmune drug, Stelara (ustekinumab). For the past few years, Stelara has been J&J’s top seller, earning $10.4 billion in 2024.
This is about reducing the time and cost and increasing the predictability of going from concept to commercialization.” – Dr. Jeff Shuren During a presentation at the May 17, 2023 Food and Drug Law Institute (FDLI) Annual Conference, CDRH Director, Dr. Jeff Shuren discussed TAP with enthusiasm.
On March 27, the FDA finalized a transition guidance document covering products that had been marketed under enforcement discretion policies during the PHE. Those concerns have proven serious enough that in September 2020 the FDA published a final guidance document calling on manufacturers of drug products to take two steps.
The developer of that treatment, Sarepta Therapeutics, informed the Food and Drug Administration his case could be life-threatening. But the Duchenne community mostly embraced Elevidys, propelling it to the fastest market launch of any gene therapy approved in the U.S. You can unsubscribe at anytime.
Research in gene therapies and genetically engineered drugs and vaccines are growing exponentially, and will only continue to become more popular. The accelerating gene therapy market is expected to grow globally by 16.6% between 2020-2027.
Multiple drugmakers agreed to cut prices of some of their newest drugs by as much as 50% in order to receive coverage from China’s national insurance program. The companies agreed to the steep price cuts in order to have their drugs made available to the world’s second-largest pharmaceutical market. billion by 2027.
RE47,739 (‘739) by more than four years until March 5, 2027. The PTE certificate was granted under the patent restoration provisions of the Drug Price Competition and Patent Term Restoration Act of 1984. Food and Drug Administration (FDA). Across clinical trials (PALOMA-1, PALOMA-2, PALOMA-3), 1.0% hypoxia, cough, dyspnea).
Sentinel Update: PDUFA projects, ARIA sufficiency and building out a method for causal inference FDA’s Sentinel Initiative, the post-market, real-world data surveillance system, is nearing the end of its current five-year strategic plan. The Sentinel Initiative was kicked off in 2008 under the FDA Amendments Act (FDAAA).
The wide range of assertions supporting or opposing Health and Human Services’ (“HHS’”) recommendation that the Drug Enforcement Administration (“DEA”) reschedule cannabis federally from schedule I to schedule III would lead an outsider to conclude that commenters are referring to different substances. Letter to U.S.
2060-AV59 Market-Based Approaches Under the National Pollutant Discharge Elimination System (NPDES) Program (Proposed Rule Stage) EPA supports market-based mechanisms to accomplish its mission to protect human health and the environment especially regarding nutrient management. Regan, Case No: 1:16-cv-00364-CRC (D.D.C.
For increased flexibility in bringing PIPs to market, a developer can also submit both. Market-based mechanisms include water quality trading under the Clean Water Act (CWA), an approach that may cost less than more traditional regulatory approaches. EPA is reviewing the comments received and is planning to issue a final rule.
Jacob Bell/BioPharma Dive Dive Brief: Moderna’s seasonal influenza vaccine met its main goal in a large Phase 3 trial, reducing the risk of influenza-like illness in people 50 years and older by 27% compared with those given a marketed shot targeting three or four strains of the virus, the company said Monday. By Jonathan Gardner • Sept.
March 2024 Greenhouse Gas Emissions Standards for Heavy-Duty Vehicles—Phase 3 (Final Rule Stage) 2060-AV50 On April 12, 2023, EPA announced a proposal for more stringent standards to reduce greenhouse gas emissions from HD vehicles beginning in model year (MY) 2027. 7521(a), starting with model year 2027.
Following the completion of the spin-off of the Upjohn Business (4) in the fourth quarter of 2020, Pfizer now operates as a focused innovative biopharmaceutical company engaged in the discovery, development, manufacturing, marketing, sales and distribution of biopharmaceutical products worldwide. in December 2019; and. in December 2019; and.
March 2024 February 2024 Greenhouse Gas Emissions Standards for Heavy-Duty Vehicles—Phase 3 Final Rule Stage 2060-AV50 On April 12, 2023, EPA announced a proposal for more stringent standards to reduce greenhouse gas emissions from HD vehicles beginning in model year (MY) 2027. 7521(a), starting with model year 2027.
Food and Drug Administration (FDA) approved Gardenia Blue, a plant-based color additive, while simultaneously making clear to industry that the Agency encourages food manufacturers to accelerate their phasing out of the use of the synthetic dye FD&C Red No. 3 in food prior to the previously announced 2027 deadline.
Deep Dive Library Events Press Releases Topics Sign up Search Sign up Search Pharma Biotech FDA Clinical Trials Deals Drug Pricing Gene Therapy An article from AbbVie to build API plant in Illinois as it steps up US production The expansion is part of AbbVie’s larger $10 billion commitment to U.S. You can unsubscribe at anytime.
The interim final ruling on most-favored-nation status for drug prices is expected to be published on November 27 th in the Federal Register by the Centers for Medicare & Medicaid Services (CMS) at the Department of Health and Huma Services (HHS). Specifically, one study of 27 drugs found the U.S. CMS maintains otherwise.
The biopharmaceutical industry is witnessing unprecedented growth, with the Contract Development and Manufacturing Organization (CDMO) market expected to reach $289.64 billion by 2027. Monitor and Adapt Marketing Strategies CDMOs must continuously monitor and adapt their marketing strategies to stay ahead of the competition.
This development is not just a significant milestone in drug discovery, but also a potential breakthrough for clinical practice, as it may address a critical gap in current treatments by offering effective stroke prevention with a reduced risk of bleeding. .
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.




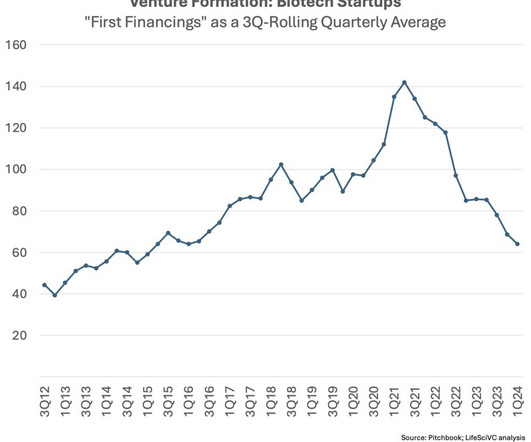




































Let's personalize your content