This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The regulatory environment in Japan for generic drug development is complex and has undergone significant changes in recent years. Regulatory Authority: Pharmaceuticals and Medical Devices Agency (PMDA) The PMDA is the primary regulatory authority responsible for overseeing the drug approval process in Japan.
For example, transcriptomic processes are showing the potential to identify and track failures in gene expression and gene regulation of amyloid and tau-related biomarkers, understood as precursors to the onset of Alzheimers disease (AD).
This partnership brings together Altasciences’ extensive expertise in preclinical research and early clinical development with VoxCell’s groundbreaking tissue engineering technology, promising to deliver a more human-relevant, predictive, and efficient drug development paradigm.
Regulatory Guidance for Oligonucleotide Bioanalysis in Drug Development pmjackson Wed, 02/19/2025 - 21:30 The unique physicochemical properties of oligonucleotides require the use of specialized bioanalytical approaches, with key considerations including selectivity and specificity, sensitivity, stability, and matrix effects.
However, as your high-school literature teacher warned youto ace the test, you need to read the book, ahem, source regulations, guidance, or other policy documents. CBERs approach here was to take FAQs from across sponsor interactions, public workshops, email requests, etc.
The FDA has recently issued this draft guidance to address clinicalpharmacology considerations for peptide drug products. These products fall somewhere between drugs and biologics and do not have the usually well-defined characteristics of either category. A peptide is any polymer with 40 or fewer amino acids.
Are Gaps in Your ClinicalPharmacology Program Jeopardizing Your Drug’s Approval? Eva Gil Berglund, PhD Justin Hay, PhD Paola Coppola, MSc Duration 60 Minutes Clinicalpharmacology information comprises more than 50% of a drug label. Click here to login. Click here to login.
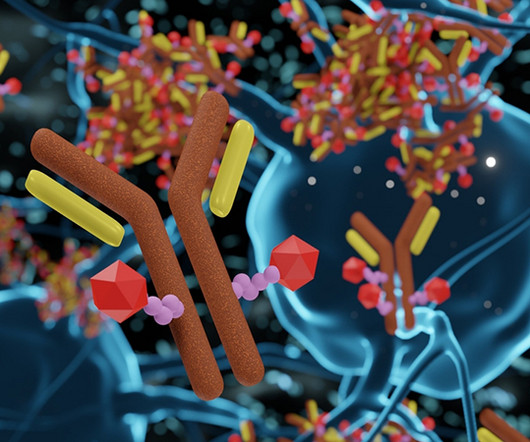
Over the last two decades, an increasing number of Antibody Drug Conjugate (ADC) therapeutics have been approved for oncology indications. These therapies have broadened treatment options for patients to expand beyond the more traditional small molecule drug alternatives. 3D rendering of Antibody Drug Conjugate Molecules.
In this blog, we explain the role of clinicalpharmacology in drug development and demonstrate how the right strategy can accelerate development under the US Food and Drug Administration (FDA) 505(b)(1) and 505(b)(2) New Drug Application (NDA) pathways.
This continued innovation highlights the complexity of the drug development process, particularly as the field is highly regulated by health authorities around the world. While it may not seem like a priority at first glance, the success of a drug can hinge on early decisions around artwork.
Nasdaq: BIIB) initiated a rolling submission of a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for zuranolone in the treatment of major depressive disorder (MDD). Zuranolone is an investigational two-week, once-daily oral drug being developed for MDD and postpartum depression (PPD).
Regulatory Excellence - Licenses for Schedule I through IV drug substances. Adherence to harmonized pharmacy-specific SOPs based on cGMP principles at our three clinicalpharmacology units. Exceptional inspection results from major regulators such as the FDA, HC, EMA, and MHRA.
3 Research Focus: Network-based drug repositioning strategy to identify drugs targeting obesity and type 2 diabetes. retrieved the drug targets from DrugBank , which they used to establish the relationship with the disease modules, finally predicting drugs that can target these modules effectively. Dutta et al.
Dr. Löbenberg holds Health Canada licences for research and analytical testing of a wide range of psychedelic compounds under the Controlled Drugs and Substance Act and research and analytical testing licences for cannabis under the Cannabis Act. .
DMT (N,N-dimethyltryptamine).
Prof. Dr. Löbenberg. ” Prof.
What We Expect the FDA to do in July and August 2024 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the months ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. This is what OIRA is currently reviewing.
FDA’s nonprescription advisors find no efficacy for phenylephrine This week, FDA’s Nonprescription Drugs Advisory Committee (NDAC) voted unanimously that current scientific data do not support the efficacy of oral phenylephrine as a nasal decongestant, aligning with FDA analysis — and re-analysis — of data.
Workshop addresses oncology dose optimization across full span of development In a series of broad-ranging, frank discussions, attendees at a joint FDA-American Association of Cancer Research (AACR) workshop worked through opportunities and challenges for dose optimization across the span of cancer drug development activities.
The 51 regulations that FDA is currently working on The FDA today unveiled its much-anticipated Spring 2023 Unified Agenda, a document outlining the regulations the agency plans to release in 2023 and beyond. The anticipated date of publication is June 2023, meaning we should see this regulation imminently.
What We Expect the FDA to do in November 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. There are as many PDUFA dates in November and December as we’ve seen all year.
What We Expect the FDA to do in June 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. An updated Unified Agenda: The Unified Agenda is the U.S.
By our count, the FDA’s Oncologic Drugs Advisory Committee held seven meetings in 2023; additionally, the Oncology Center of Excellence authored nine new (draft) and six final guidance documents. As more data have been collected on products previously approved, regulators have started to look back at them with a more critical gaze.
FAR regulations require contracting officers to “promote and provide for full and open competition in soliciting offers and awarding Government contracts.” The assessment should include recommendations “on further enhancements” for hiring and retention of staff in FDA’s human drug review program. FDA has previously worked with ACMT.
A closer look at CDER’s new 2024 guidance agenda The FDA’s Center for Drug Evaluation and Research, its drug review division, this week quietly updated its 2024 Guidance Agenda, a list of all draft guidance documents the agency is working on this calendar year. This list is known as the FDA’s “guidance agenda.”
10 June 2022 Language EN Drug repurposing is a strategy for identifying new uses for approved drugs, outside the scope of the original indication. Below, we have listed recent findings about the repurposing of generic drugs in oncology. It is one of the focus areas of the Anticancer Fund.
What We Expect the FDA to do in May and June 2024 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the months ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. This is what OIRA is currently reviewing.
.” Interchangeable products can be substituted for the reference product at the pharmacy level, without prescriber intervention, within state and local regulations. The size of the trials was comparable to those of new molecular entities—drugs that have never before been approved by the FDA. million (IQR: $18-36.7 million). “We
How Improving Diversity Can Benefit Clinical Trials pmjackson Wed, 07/31/2024 - 19:19 In July 2024, the U.S. Food and Drug Administration (FDA) published a draft guidance to ensure greater diversity in clinical trials, which is expected to become a final guidance by June 2025.
What We Expect the FDA to do in October 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. AgencyIQ October 1 Nitrosamine testing due to FDA.
FDA updates guidance on developing drugs for Covid-19, replacing pandemic-era version Last week, FDA published the third update to its guidance on the development of products to prevent or treat Covid-19. The FDA also implemented new flexibilities for certain regulated products and processes, typically via enforcement discretion.
What We Expect the FDA to do in December 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. December also typically brings end-of-year perspectives on the agency’s accomplishments.
What We Expect the FDA to do in February and March 2024 (Updated) In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the months ahead, including key deadlines, meetings, events, planned regulations, comment periods and more.
What We Expect the FDA to do in March and April 2024 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the months ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. Read AgencyIQ’s analysis of a lower court’s ruling.
Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. Thyroid replacement therapy, when clinically indicated, should be initiated or dose-adjusted. DRUG INTERACTIONS. Drug Interactions – Based on in vitro data, ZYTIGA ® is a substrate of CYP3A4.
The 53 regulations that FDA is currently working on The FDA on Friday unveiled its much-anticipated Spring 2024 Unified Agenda, a document outlining the regulations the agency plans to release in 2024 and beyond. Of those 53, seven are new to the Agenda, having never before been included in prior agendas.
Introduction Biomarkers are becoming increasingly essential in drug development and clinical practice, driving the need for more precise validation methods. 1 The journey to qualifying biomarkers for clinical and regulatory use is fraught with challenges, leading to a remarkably low success rate. For example, only about 0.1
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






































Let's personalize your content