This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
It is recognized, however, that there is a learning curve across the industry and FDA that is inherent to the development of such novel technologies. To prevent this learning curve from becoming a roadblock on the path to regulatory approval, the FDA has developed several collaborative programs to support advanced manufacturing.
Farquhar This is the first in a series of blog posts on tips for successfully handling an FDA inspection. Using publicly available examples, these lessons will illustrate potential pitfalls and strategies for interacting with FDA during and after an inspection. FD&C Act 501(j).
But growing ethical scrutiny, supply shortages, cost burdens, scientific innovation and regulatory shifts like the US Food and Drug Administration (FDA)’s new alternative methods roadmap are bringing the continued reliance on live NHPs into question, and opening the door to next-generation solutions that could eventually replace them altogether.
FDA Approves Merck’s ENFLONSIA™ to Protect Infants from Severe RSV Illness Merck operating as MSD outside the United States and Canada, has received a significant regulatory milestone with the U.S. Food and Drug Administration (FDA) granting approval for ENFLONSIA™ (clesrovimab-cfor). A New Era in Infant RSV Protection Dr. Dean Y.
& Researchers at the Broad Institute and The Jackson Laboratory have used prime editing, a precise and versatile form of gene editing, to correct the root cause of AHC in mice. There is no cure or effective treatment for this rare genetic disease, but new research suggests a potential path to one.&
These standards are widely recognized by regulatory agencies such as the FDA and EMA for use in analytical method development, validation, and quality control.
Clinical research generates vast amounts of diverse data from laboratory tests, patients, medical equipment, and outside sources. By organising and analysing this information, researchers can extract actionable insights that improve patient outcomes, data accuracy, drug efficacy and speed up trials.
Based on a technology developed by Broad Institute core member David Liu’s laboratory, the treatment is the first in a series of new medicines being tested to treat rare diseases by repairing patients’ particular genetic misspellings. The team that treated K.J. This unprecedented feat required diagnosing K.J.’s
Jayaprakash Kotha, MBBS, PhD, ASCP (SH), Vice President, Bioanalytical Laboratory Satish Kumar, MBB, Head of Process Improvement Continuous Innovation is a Cornerstone of Bioanalysis Approximately 80% of drugs that begin the research process fail to reach approval. What is one contributing factor that sets the 20% that do apart from the rest?
Following clear processes, in accordance with GLP and current FDA/EMA guidelines—and supported by our unique array of platforms and large list of validated assays —our team ensures the most effective methods for your individual programs. We have state-of-the-art, purpose-built laboratories at our locations in the U.S.
FDA Grants Accelerated Approval to Regeneron’s Lynozyfic™ (linvoseltamab-gcpt) for Relapsed or Refractory Multiple Myeloma Regeneron Pharmaceuticals has secured a critical milestone in oncology drug development with the U.S.
Food and Drug Administration (FDA) granted approval to ENFLONSIA in early June 2025 based on compelling results from two pivotal clinical trials: the Phase 2b/3 CLEVER trial and the Phase 3 SMART trial. Dr. Richard M. Clinical Evidence Supporting ENFLONSIA The U.S.
Working at the famed Pasteur Institute, the duo began by figuring out how best to culture mycobacteria — a seemingly simple, but essential first step, given how no one at the time had yet come up with an effective way of growing these bacteria in the laboratory. coli bacteria from growing. Can We Lay This Scourge to Rest?
The treatment, now known as Casgevy, became the first CRISPR-based therapy to gain FDA approval, in 2023. Notably, this has led to the development of new medicines to treat genetic diseases — Casgevy was the first of these to gain FDA approval and is used to treat two blood disorders, called sickle cell disease and beta thalassemia.
Currently, three FDA-approved disease-modifying drug therapies are available: hydroxyurea, crizanlizumab and L-glutamine, though each has limitations that affect patient compliance. The use of FDA-approved medications for preventing vaso-occlusive events in sickle cell disease. JAMA Netw Open. 2023 Nov 1;6(11):e2344546. 2023.44546.
FDA Grants Priority Review to Merck’s WINREVAIR™ Based on Landmark ZENITH Trial Showing Dramatic Reduction in Morbidity and Mortality in PAH Patients Merck (NYSE: MRK), operating as MSD outside the U.S. The FDA has set a Prescription Drug User Fee Act (PDUFA) target action date of October 25, 2025.
In 2017, small and medium-sized biotech companies accounted for 51% of FDA market approvals, while large pharma companies were the originators in only 28% of approvals[4]. A CDMO should have a mature data integrity program in place, especially in the laboratory.
FDA Approves KEYTRUDA® (Pembrolizumab) for Perioperative Treatment of Resectable Locally Advanced Head and Neck Squamous Cell Carcinoma Merck known as MSD outside the United States and Canada, recently announced that the U.S.
” Nobody really knows without trying it out in the laboratory. For one, life itself grows and develops slowly, and the many steps required to “do” biological research in the laboratory are often tedious and manual. Even in vitro experiments — or those done in the laboratory — are relatively slow because E.
Food and Drug Administrations (FDA) diversity and inclusion in clinical trials mandate. PPD Laboratory services Central Lab offers an integrated, flexible, one-stop solution for the collection, management and analysis of lab and study data in the clinical trial ecosystem.
Published June 26, 2025 Ned Pagliarulo Lead Editor post share post print email license An Incyte researcher works in a laboratory. questions vaccine evidence By Delilah Alvarado FDA investigating Elevidys safety; Nektar shares spike on eczema data By BioPharma Dive staff Cassidy challenges RFK Jr. You can unsubscribe at anytime.
Michael Laposata and the Association for Molecular Pathology in the recent LDT litigation, the Federal, Food, Drug, and Cosmetic Act does not authorize FDA to regulate LDTs as medical devices. Just before this decisive event, though, FDA released a relatively rare Warning Letter to a manufacturer of research use only (RUO) reagents.
Jordan ordered that FDAsLaboratory Developed Tests (LDT) Final Rule be vacated and set aside, in its entirety. Their case was later consolidated with a similar lawsuit filed by the American Clinical Laboratory Association (ACLA) and HealthTrackRX Indiana, Inc. By Steven J. Gonzalez On March 31, 2025, U.S.
Lenz, Principal Medical Device Regulation Expert Last year, FDA issued a letter to the medical device industry warning medical device firms of concerns related to fraudulent and unreliable laboratory testing data in premarket submissions, which we blogged about here. By Adrienne R.
Sasinowski On December 5, 2024, FDA published a new draft guidance on accelerated approval providing a much needed and substantial update to its guidance on the pathway. FDAs application and use of accelerated approval has evolved dramatically since it was first developed by the Agency to help address the HIV/AIDS epidemic in the late 1980s.
Over two decades ago, as a NIDA grantee, I and my colleagues at Brookhaven National Laboratory and SUNY-Stony Brook used PET neuroimaging to show the recovery of lost dopamine transporters in the striatum of people with methamphetamine use disorder after prolonged abstinence.
Limited Evidence for Nalfmefene "In 2021, due to the widespread availability of high-potency synthetic opioids like fentanyl, the US FDA approved two high-dose naloxone products, an 8 mg IN spray (Kloxxado) and a 5 mg IM injectable (Zimhi). In 2023, the FDA approved a 2.7 mg IM vs. 8 mg IN and 2 mg IM vs. 5 mg IM, respectively).
Baumhardt, Principal Medical Device Regulatory Expert In January 2025, FDA posted the 2024 Annual Report concerning the Accreditation Scheme for Conformity Assessment (ASCA) program as required by Medical Device User Fee Amendments of 2017 (MDUFA IV).
This commitment represents one of the most significant matching donation campaigns ever undertaken in the biopharmaceutical industry and signals a deepened investment in improving patient outcomes beyond the laboratory or clinic. Food and Drug Administration (FDA). A Philanthropic Commitment Grounded in Equity Leonard Schleifer, M.D.,
Gibbs The multi-decade battle over FDAs power to regulate Laboratory Developed Tests (LDTs) had its day in court earlier this week. As our readers will recall, the LDT rule will bring, by FDAs estimates, hundreds of thousands of laboratory tests under FDAs regulatory purview. Mullen & Jeffrey N.
Food and Drug Administration (FDA) within 30 days, providing a structured timeline for progressing with clinical trials. Early engagement with the FDA can help set expectations for future regulatory steps and ensure that non-clinical and clinical trial data meet global regulatory requirements.
The HER3-targeting treatment could become the next antibody-drug conjugate to emerge from Daiichi Sankyo’s laboratories, after the AstraZeneca-partnered Enhertu.
MONDAY, April 29, 2024 -- Laboratory tests used by millions of Americans are soon to be classified as medical devices, and as such be regulated by U.S. Food and Drug Administration, the agency announced Monday.The new rule does not apply to tests.
Walsh — Among FDA-regulated establishments and stakeholders, there is one word that makes everyone go on edge – the dreaded FDA “inspection.” In FY2023, FDA conducted over 1000 inspections under the BIMO program. We also detail some of our recommended best practices to achieve success when FDA comes knocking.
Mullen — On January 31, 2024, FDA announced its intent to initiate the reclassification process for most in vitro diagnostic (IVD) products that are currently class III (high risk) into class II (moderate risk). This is to say nothing of the expected doubling of annual device submissions FDA expects it will then receive on an ongoing basis.
A Texas judge vacated the FDA’s final rule, which was strongly opposed by the laboratory industry, and remanded the matter to HHS Secretary Robert F. Kennedy Jr.
FDA set to unveil new rule on Laboratory Developed Tests this August Following challenges getting a diagnostics reform package through Congress, known as the VALID Act, the FDA has just announced that it plans to release a proposed rule in August 2023 that would change the way that the agency effectively regulates laboratory developed tests (LDTs).
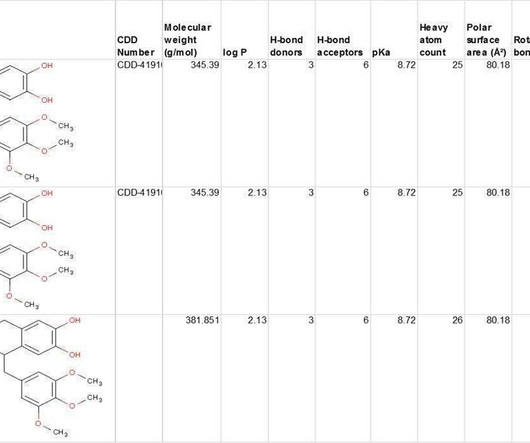
CDD Vault provides conventional SAR tables of course, but it also gives you access to data from multiple public sources for comparison with hundreds of published sources, including popular MLSMR, GlaxoSmithKline TCAMs, and FDA-Approved Re-purposed Drugs data sets.
The webinar largely consisted of summarizing the general requirements under Parts 803, 806 and 820.198, which we do not reproduce here ( but see another of our prior blog posts discussing these requirements and their applicable to LDTs in greater detail; you can also find FDA’s slides from the webinar here ).
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
On May 6, 2024, the US Food and Drug Administration (FDA) issued the Final Rule regarding regulation of laboratory developed tests (LDTs). The LDT Final Rule outlines a four-year phaseout period for the enforcement discretion approach that FDA has previously exercised for LDTs.
Mullen — On January 18, 2024, the director of FDA’s Center for Devices and Radiological Health and the chief medical officer and acting director of CMS’ Center for Clinical Standards and Quality issued a joint press release supporting FDA’s recent proposed rule regulating Laboratory Developed Tests (LDTs).
Gibbs — On March 21, 2024, the House Energy and Commerce held a subcommittee hearing titled “Evaluating Approaches to Diagnostic Test Regulation and the Impact of the FDA’s Proposed Rule.” They believe any regulatory oversight for laboratory developed tests (LDTs) should be mandated by Congress, rather than the Executive Branch.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

















































Let's personalize your content