This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This technique " opens large windows into the cell’s interior ," according to a 2013 review, allowing visualization of hidden structures that can’t be easily extracted or crystallized. Today, a single injection of an FDA-approved gene therapy, called Hemgenix , cures this disease. Credit: SIngh U.S.
A: This is a complex topic and is best outlined in the FDA’s guidance document. Q: What is the typical time period between the submission of the briefing package and the pre-IND meeting? A: The FDA’s guidance document indicates the briefing package is submitted four weeks before the meeting. of this guidance.
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Through this program, the FDA can conduct periodic stability testing on select products and extend the labeled shelf life accordingly.
Within the realm of FDA-required labeling, there are currently a few different types of information a sponsor might develop specifically for patient use: medication guides, instructions for use (IFU), consumer medical information (CMI) and patient package inserts (PPI). After all, companies already have FDA-approved labels.
IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. IMBRUVICA was one of the first medicines to receive FDAapproval via the Breakthrough Therapy Designation pathway. 8 VENCLEXTA (venetoclax) [Package Insert].?North?Chicago, BTK signaling is needed by specific cancer cells to multiply and spread.
IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. IMBRUVICA was one of the first medicines to receive FDAapproval via the Breakthrough Therapy Designation pathway. 9 VENCLEXTA (venetoclax) [Package Insert].?North BTK signaling is needed by specific cancer cells to multiply and spread. 8 IMBRUVICA U.S.
The FDA requires real science for warnings; thus it had not mandated any warning remotely resembling Prop 65. The plaintiff failed to identify any method by which a generic (or any other) drug manufacturer could add a Prop 65 warning without deviating from FDA-approved labeling, thereby violating federal law. 13) McGee v.
The case alleged on-label drug use between 2009 and early 2012, purportedly leading to plaintiffs’ decedent’s suicide in 2013 – more than a year after use of the drug had ceased. Both propositions are well recognized, but Pfaff ties them together in one neat package.
472 (2013), had not arrived and plaintiffs had not stopped suing over alleged injuries from generic drugs. To the contrary, as one of the panel noted in a concurrence, FDAapproval of a drug’s labeling creates a presumption of adequacy. Plaintiffs did not even complain about the content of the package insert.
2015), finally gave appellate recognition to the preemption of design defect claims for FDA-approved branded prescription drugs. FDAapproved the drug with its particular formulation and the manufacturer could not have changed the formulation on its own. Ortho-McNeil-Janssen Pharms., 3d 281 (6th Cir. Bartlett , 570 U.S.
13] [14] The first regulatory approval was granted in Germany in August 1999 to Madaus AG for Regurin 20 mg Tablets. [15] 15] :13 Madaus licensed the US rights to trospium chloride to Interneuron in 1999 and Interneuron ran clinical trials in the US to win FDAapproval. [17] November 2013). billion in 2004. [23]
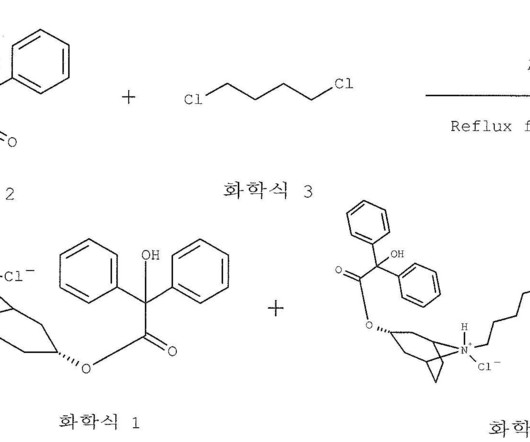
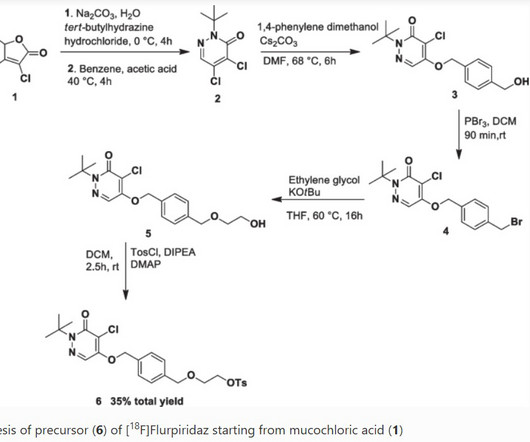
3] Flurpiridaz ( 18 F) was approved for medical use in the United States in September 2024. [3] Jump up to: a b c d e f g h i j k “FDAapproves imaging drug for evaluation of myocardial ischemia” U.S. Food and Drug Administration (FDA). Food and Drug Administration (FDA). 2013 Jan 29;61(4):469-477.
Further, the pace of fast follower drugs is striking relative the past: according to PhRMAs recent report on innovation , the average time for a drug class to have three FDAapprovals in it went from ~15 years in 1990-2003 to ~2 years in 2013-2021.
Zidovudine showed promise against multiple HIV strains in cultured cells, and the Food and Drug Administration (FDA) approved it for human studies within five months. By 1987, the FDA licensed zidovudine after trials showed it increased survival rates.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



















Let's personalize your content