This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Daily Dive M-F Commercialization Weekly Every Wednesday Gene Therapy Weekly Every Thursday Emerging Biotech Weekly Every Tuesday By signing up to receive our newsletter, you agree to our Terms of Use and Privacy Policy. subsidiary of Merck KGaA, is the founder of radioligand therapydeveloper Actithera.
As our understanding of the underlying biology of disease grows more sophisticated, emerging therapies operate on increasingly complex biopathological systems and mechanisms. Safety biomarkers account for adverse effects of a therapy under study. There are several types of biomarkers to consider.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
FDA Orphan Drug Designation for Antibody-Mediated Rejection in Solid Organ Transplantation The U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation (ODD) to riliprubart , an investigational immunology therapydeveloped by Sanofi, for the treatment of antibody-mediated rejection (AMR) in solid organ transplantation.
They have interesting patterns of expression in different cancer indications; thus, diverse therapies for attacking these targets have been developed. Each target and each therapeutic modality induce varying degrees of clinical efficacy, as well as causing toxicities. months (versus 20% ORR and 8.3
Researchers must characterize the anti-drug-antibody (ADA) response in preclinical and clinical studies and report any ADA-positive samples as a risk-based approach. Regulatory Considerations for Oligonucleotide Drug Development and Safety In 2024, the U.S.
This design was meant to enable clinician-researchers to gauge the ability of rilzabrutinib to control disease activity after the withdrawal of standard therapy. We look forward to further evaluating rilzabrutinib in subsequent phases of its clinicaldevelopment.” Fast Track and Orphan Drug Designations from FDA The U.S.
Approximately two in 10 patients with hematologic malignancies are estimated to be eligible and able to receive cell therapy. The REMS Burden Explained Once deemed necessary for managing the risks associated with CAR T therapies, the REMS requirements imposed significant operational burdens on treatment centers.
(Nasdaq: IONS) has announced the dosing of the first patient in the Phase 3 REVEAL clinical trial, marking a significant milestone in the development of ION582, an investigational therapy for Angelman syndrome (AS).
Commitment to Innovation in Rare Blood Disorders Sanofi’s pipeline, centered on the complex biological mechanisms that drive rare blood diseases, reflects a strategic investment in therapies that aim to address persistent unmet medical needs. and EU, in late 2024 as the first therapy of its kind. In the U.S.,
Drug development is a complex and highly regulated process. Before a therapy can be approved for patient use, it must undergo extensive clinical testing and strictly adhere to regulatory guidelines. A well-documented case is Sarepta Therapeutics’ eteplirsen , a drug developed for Duchenne muscular dystrophy.
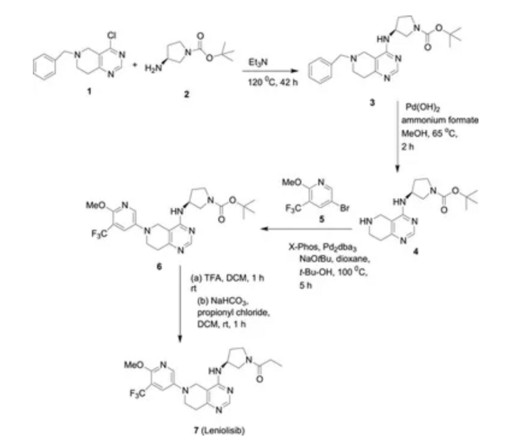
5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [9] 5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [9] Jump up to: a b c d e f g h i j “FDA approves first treatment for activated phosphoinositide 3-kinase delta syndrome” U.S.
Daily Dive M-F Commercialization Weekly Every Wednesday Gene Therapy Weekly Every Thursday Emerging Biotech Weekly Every Tuesday By signing up to receive our newsletter, you agree to our Terms of Use and Privacy Policy. Among those who had gotten another such therapy, the rates were 52% and 62% in those trials.
FDA Grants Priority Review to Merck’s WINREVAIR™ Based on Landmark ZENITH Trial Showing Dramatic Reduction in Morbidity and Mortality in PAH Patients Merck (NYSE: MRK), operating as MSD outside the U.S. The FDA has set a Prescription Drug User Fee Act (PDUFA) target action date of October 25, 2025. risk score of 9 or greater.
They have interesting patterns of expression in different cancer indications; thus, diverse therapies for attacking these targets have been developed. Each target and each therapeutic modality induces varying degrees of clinical efficacy, as well as causing toxicities. These activities are pro-oncogenic (reviewed here: [link] ).
The new recommendation supports the use of darolutamide in combination with androgen deprivation therapy (ADT) for patients with metastatic hormone-sensitive prostate cancer (mHSPC) in the European Union (EU). Food and Drug Administration (FDA) already approved darolutamide in combination with ADT for mHSPC in June 2025.
FDA Approves KEYTRUDA® (Pembrolizumab) for Perioperative Treatment of Resectable Locally Advanced Head and Neck Squamous Cell Carcinoma Merck known as MSD outside the United States and Canada, recently announced that the U.S. Subsequently, KEYTRUDA is used as monotherapy following the completion of adjuvant therapy. Source link
Grifols’ BT524 Fibrinogen Therapy Shows Promise in Phase 3 Trial Published in The Lancet’s eClinicalMedicine Grifols , a globally recognized healthcare company and a leader in the production of plasma-derived therapies, has marked a significant advancement in the treatment of acquired fibrinogen deficiency (AFD).
Valentine On November 19, 2024, FDA released a draft guidance titled Frequently Asked Questions Developing Potential Cellular and Gene Therapy Products. Section #1: FDA Interactions Given the wide range of sponsors (i.e., Tobolowsky & Charles G. Raver & James E.
They wait for the therapies that could significantly improve their quality of life or even save it knowing approval could be years or decades away. And yet, despite this learning, traditional clinicaldevelopment processes have failed to evolve; they still have high asset failure rates, longer development timelines, and significant costs.
Daily Dive M-F Commercialization Weekly Every Wednesday Gene Therapy Weekly Every Thursday Emerging Biotech Weekly Every Tuesday By signing up to receive our newsletter, you agree to our Terms of Use and Privacy Policy. You can unsubscribe at anytime. Since the drug’s U.S.
It is currently in Phase III clinicaldevelopment by the German biopharmaceutical company AiCuris Anti-infective Cures AG. US FDA granted fast track designation for pritelivir in 2017 and breakthrough therapy designation 2020. This is particularly important in immune compromised patients.
Valentine We recently published the first part of our review of FDAs draft guidance titled Frequently Asked Questions Developing Potential Cellular and Gene Therapy Products. If you make a tissue product you may produce one lot per patient where a gene therapy may only need a handful of lots through Phase 3.
Daily Dive M-F Commercialization Weekly Every Wednesday Gene Therapy Weekly Every Thursday Emerging Biotech Weekly Every Tuesday By signing up to receive our newsletter, you agree to our Terms of Use and Privacy Policy. You can unsubscribe at anytime.
This expansion is creating opportunities for clinical trials related to a range of new therapy areas and their subpopulations. Food and Drug Administrations (FDA) diversity and inclusion in clinical trials mandate. It is worth noting that trial ran from 2018 to 2021, before the U.S.
ML is also used to identify predictors of response to a therapy or risk for adverse events (AEs) that can be used to inform value-based contracting or treatment strategies intended to minimize “wasted” use of a drug. Ready to learn more about convergence of real-world data and technology for clinical trials?
Daily Dive M-F Commercialization Weekly Every Wednesday Gene Therapy Weekly Every Thursday Emerging Biotech Weekly Every Tuesday By signing up to receive our newsletter, you agree to our Terms of Use and Privacy Policy. You can unsubscribe at anytime. Thus, these drugs best complement, rather than replace, healthy lifestyle interventions.
The FDA has approved a request from American Gene Technologies to begin a clinical study into its HIV gene therapy. In a paper published in Molecular Therapy: Methods & ClinicalDevelopment , the team discussed why they choose this type of gene therapy to treat HIV. Conor Kavanagh. Source link.
The field of cell and gene therapies (CGT) is constantly evolving, and there has been significant progress in this area of research. However, despite the promise of these therapies, the regulations governing them lag the science, which in turn hinders the clinical translation of these novel medicines.
and Shionogi Limited as shareholders, today announced the positive findings of a pooled analysis of COVID-19-related impacts across the investigational long-acting cabotegravir and rilpivirine clinicaldevelopment programme. Of those participants who transitioned back to injectables, the median duration of oral therapy was 51 days.
Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.” Tobolowsky & Richard A. It finalized a draft guidance published in 2022.
Read the Guide FDA Launches Pilot Program to Help Further Accelerate Development of Rare Disease Therapies One frustrating aspect of traditional drug development, especially for rare disease communities, is the tempo of regulatory decisions on potential drugs.
Brevig, Senior Regulatory Device and Biologics Expert — On December 7, 2022, FDA’s Center for Biologics Evaluation and Research (CBER) and the Office of Tissues and Advanced Therapies (OTAT) held a town hall to answer questions related to cell therapy and tissue-engineered products chemistry, manufacturing, and controls (CMC).
Food and Drug Administration (FDA) for the treatment of adult patients with deleterious or suspected deleterious BRCA -mutated ( BRCA m) metastatic castration-resistant prostate cancer (mCRPC). Patients should be selected for therapy based on an FDA-approved companion diagnostic for LYNPARZA. In the U.S., In the U.S.,
. – Preclinical Data Underscore Treatment Potential for PBFT02 in Frontotemporal Dementia with Granulin (GRN) Mutations, a Devastating, Progressive Disorder Impacting Adults with No Approved Disease-Modifying Therapy Options. FTD is a debilitating form of early onset dementia that currently has no approved disease-modifying therapies. “We
Contrary to popular belief, ageing is not caused by just random wear and tear of our bodies over time but is instead caused by a discrete set of biological mechanisms that we now better understand and can target with therapies. a clinical-stage private biopharma company developingtherapies for neurological and psychiatric diseases.
Even though AI-designed drugs arent yet a household term for FDA-approved, commercially available therapies, they are a reality in clinicaldevelopment pipelines.
Data is the cornerstone of any clinical trial, driving the decision-making process of drug development, and is a fundamental requirement for approval of new therapies by regulatory agencies like the FDA or EMA.
The US Food and Drug Administration (FDA) has accepted Roche’s application for a new self-administration option for Xolair (omalizumab) across all approved US indications. Its use in these settings is supported by a rigorous clinicaldevelopment programme, including eight Phase III studies.
Source link.
The FDA’s January 2020 guidance, Chemistry, Manufacturing and Control (CMC) [1] Information for Human Gene Therapy Investigational New Drug Applications (INDs), outlines the analytical methods that define the quality, safety and efficacy of gene therapy therapeutics.
Food and Drug Administration (FDA) granted Breakthrough Therapy Designation (BTD) to TAK-994, 1 its Phase 2 investigational oral orexin agonist, which is designed to selectively target orexin 2 receptors. – If Approved, Investigational TAK-994 May Provide a Future Treatment Option Targeting the Orexin Deficiency Underlying NT1.
BY RACHEL COE, MSC | FEB 7, 2024 10:21 PM CST The Bespoke Gene Therapy Consortium A collaborative effort, the Bespoke Gene Therapy Consortium (BGTC) was launched in October 2021 to “accelerate development of gene therapies for the 30 million Americans who suffer from a rare disease.”
today announced a label update for KEYTRUDA, Merck’s anti-PD-1 therapy, for its indication in first-line advanced urothelial carcinoma (bladder cancer) in the U.S. Food and Drug Administration (FDA) has converted this indication from an accelerated to a full (regular) approval. Source link: [link].
While some clinical trials are necessary to confirm safety and efficacy in humans, these are typically smaller and shorter in duration compared to the extensive trials required for novel biopharmaceuticals. Food and Drug Administration (FDA) have been instrumental in shaping global regulatory frameworks for biosimilars.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content