This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The distinction between it and traditional corticosteroid drugs lies in its capacity to specifically activate particular signaling pathways of corticosteroids. In individuals diagnosed with DMD, the primary mechanism through which corticosteroid drugs exhibit their effectiveness is by exerting anti-inflammatory effects. 70) Keam, S.
Published June 24, 2025 Gwendolyn Wu Senior reporter post share post print email license Lexeo Therapeutics and two life sciences investors are working together to launch a startup to develop cardiac RNA therapies. By Jonathan Gardner • Sept.
Published July 14, 2025 Ben Fidler Senior Editor post share post print email license Bain Capital, Kohlberg and Mubadala joined to invest in biopharmaceutical CDMO PCI Pharma on July 14, 2025. PCI helps biopharmaceutical companies manufacture and packagedrug products used in clinical trials as well as commercially.
Originally developed in the 1970s to treat diabetes, these drugs—such as Ozempic, Wegovy, and Mounjaro—have become headline-makers for their ability to induce significant weight loss. Understanding GLP-1 drugs GLP-1, or glucagon-like peptide-1 receptor modulators, mimic natural hormones that regulate insulin and appetite.
This product was developed by uniQure prior to being licensed to CSL Behring. Of course, neither “cure” nor permanency are the bar for FDA to find that a new drug is effective; simply because there is the potential for something to be a “cure” or offer permanent/near-permanent effects does not mean it should be required to be such.
Butler — In a move FDA is calling “radical transparency,” the Agency announced on July 10, 2025 that it has published 200+ Complete Response Letters (CRLs) issued in response to marketing applications for drugs and biologics on its openFDA database.
In this example, we'll be using the chemfp package by Andrew Dalke. Chemfp is a great package. If you're using it for production drug discovery, you should buy a license. Chemfp has both free and paid tiers. With the free tier, you can perform similarity searches on smaller datasets, like the one we're using here.
Determining if you have a combination product The high-level definition of combination product seems straightforward: a product with at least two constituent parts, such as: Drug/biologic (e.g., therapeutic drug/monoclonal antibody) Drug/device (e.g., insulin injector pen) Multiple drugs/device (e.g.,
On July 31, 2024, the US Food and Drug Administration (FDA) announced Fiscal Year 2025 (FY2025) Prescription Drug User Fee Amendments of 2022 (PDUFA VII) fee rates for the review of human drug and biological product applications along with prescription drug program fees.
These features should not be included on outer packaging or immediate packaging, if there is no outer packaging. Last week, the MHRA provided a roadmap for upcoming labeling requirements, based on license type The Windsor Framework allows the MHRA to approve medicines for U.K. wide license: The requirements of the E.U.
But commitment to neuroscience drug development is critically important given the significant unmet medical needs and the ways in which patients, caregivers, and families suffer. Not failed drugs, but failed studies. As soon as drug developers can begin to develop conviction, momentum increases.
If the sponsor wants their drug approved, they need to complete all clinical studies and submit an application. Q: Please discuss the transfer of investigational new drug (IND) sponsorship from one sponsor to another and that process. Q: Can you ship a drug from another country to the U.S. A: Yes, the EUA is just temporary.
One of the most common formulation issues that sponsors of new drug or biologic products face is the qualification of novel excipients. Therefore, when excipients are selected for formulating a new drug product, it is essential that they either meet current FDA standards or are qualified for use in the drug product.
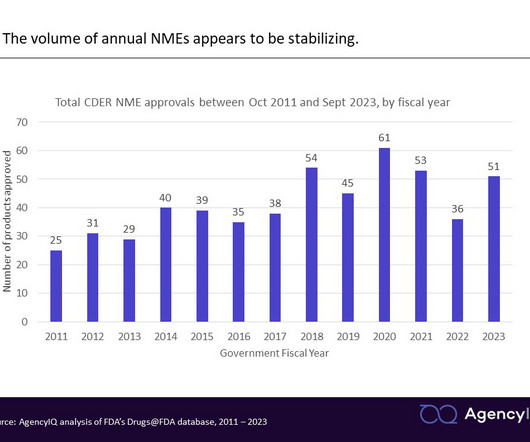
CDER’s latest novel drug approvals report shows how the pandemic is still affecting some drug approvals According to a report released this week describing CDER’s approval of 55 novel drugs in 2023, the agency failed to meet its goal dates under the Prescription Drug User Fee Act (PDUFA) Commitment Letter for 11% of applications – a record high.
The addition of three medial-to late- stage clinical programs into our oncology channel significantly expands the number of transformational drugs we can potentially deliver to people with cancer, while also enabling our pursuit of new combinations.”. Terms of the Exercised Options and Amendment to the Agreement. About Gilead Lores.
Mr. Barrow has over a decade of experience leading drug development programs aimed at identifying and testing novel treatments in a wide range of disease conditions under FDA and EMA.
We are excited to attract such top tier talent from the psychedelic drug development community. .
NEW YORK , Jan.
AbCellera’s AI-powered antibody discovery platform speeds the otherwise lengthy and grueling process by analyzing the database of natural immune systems to find antibodies that can be developed into drugs. Tackle the toughest problems in drug development.” The promise to partners is to “move quickly. Reduce cost. Remix Therapeutics .
As has been explained, the BPCIA provided an abbreviated pathway to market for follow-on biologics—biosimilars and interchangeable biosimilars—and with that pathway came two types of exclusivity periods: one for the “Reference Product” to encourage continued innovation and one for the “First Interchangeable” biosimilar to encourage the development (..)
Food and Drug Administration (FDA)’s recent agreement to allow the NexoBrid expanded access (NEXT) protocol to be expanded to include pediatric as well as adult burn patients. “We patients during the review of the NexoBrid Biologics License Application (BLA). Additionally, the completion of the CIDS enrollment stage follows the U.S.
These statements concern, and these risks and uncertainties include, among others, the impact of SARS-CoV-2 (the virus that has caused the COVID-19 pandemic) on Regeneron’s business and its employees, collaborators, and suppliers and other third parties on which Regeneron relies, Regeneron’s and its collaborators’ ability to continue (..)
Protocol and amendments Purpose: To document revisions of trial-related documents during the trial Documents: Log of protocol changes, IRB-approved protocol with principal investigator (PI) signature page, IRB-approved case report forms, IRB-approved advertisements and participant information sheets, protocol amendments Informed consent documents Purpose: (..)
Food and Drug Administration , which is currently evaluating REGN-COV2 for a potential Emergency Use Authorization in mild-to-moderate outpatients at high risk for poor outcomes. Finally, the IDMC recommends continuation of the outpatient trial without modification. Regeneron is also informing the U.S.
FDA Regulations and guidance under OIRA review as of July The White House’s Office of Information and Regulatory Affairs (OIRA) is the regulator of regulators, tasked with ensuring that all federal policies and regulations adhere to laws, existing regulations, federal policies and the wishes of the President.
Tonix is a clinical-stage biopharmaceutical company focused on discovering, licensing, acquiring and developing small molecules and biologics to treat and prevent human disease and alleviate suffering. TNX-102 SL, TNX-601 CR, TNX-1600 and TNX-1900 are investigational new drugs and have not been approved for any indication.
Food and Drug Administration (FDA) for the treatment of mild to moderate COVID-19 in patients 12 years of age and older and weighing at least 40 kg, who have received positive results of direct SARS-CoV-2 viral testing and are at high risk for progressing to severe COVID-19 and/or hospitalization.
Happy 40 th Birthday, generic drugs: September 24 will mark the 40 th anniversary of the 1984 signing of the Hatch-Waxman Act, which created a formal pathway for the approval of generic drugs. We expect the FDA to mark the occasion, especially since drug pricing continues to be such a potent issue in the Presidential election.
“The first job of an antiviral therapeutic drug is to lower the viral load, and our initial data in 275 patients strongly suggested that the REGN-COV2 antibody cocktail could lower viral load and thereby potentially improve clinical outcomes. . Regeneron has shared these results with the U.S.
About the Phase 1/2 Dose-escalation Trial REGN5458 monotherapy is being investigated in an open-label, Phase 1/2 dose-escalation trial in patients with R/R multiple myeloma who are at least triple refractory to existing therapeutic options, including proteasome inhibitors, immunomodulatory drugs and CD38 antibody treatments.
This results in more fully packaged and intact capsids, which are essential for reducing manufacturing costs per dose and enhancing the effectiveness of gene therapies. Kailera is one of at least six companies formed since the start of last year that were built around drugs originating from China. You can unsubscribe at anytime.
FDA fiscal year in review: New drug approvals in the wake of the pandemic and legislative reforms AgencyIQ analyzed CDER’s novel drug approvals in Fiscal Year 2023, identifying a recovery in approval numbers as the agency resumes a new normal following the pandemic. As a reminder, the U.S.
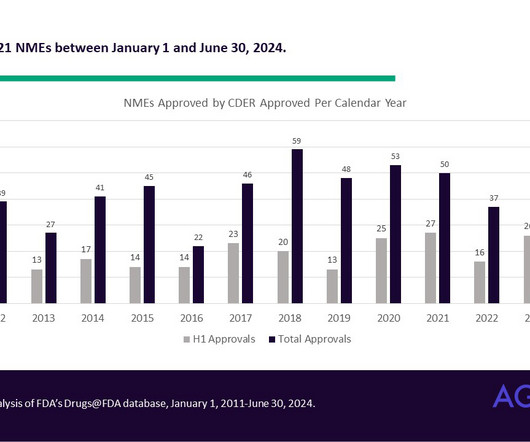
Halfway there: Novel drug approvals and their supportive clinical trials so far in 2024 In the first half of 2024, the FDA’s Center for Drug Evaluation and Research (CDER) approved 21 novel drug products. Novel drug products are defined as products that have never been approved for any indication.
Perhaps the most important addition to FDA’s agenda is a proposed rule intended to clarify that Laboratory Developed Tests (LDTs) are to be regulated as medical devices under the Federal Food, Drug and Cosmetic Act (FD&C Act). Read our analysis of that rule here and here. ]
ASHP’s full suite of drug information has been integrated into and made available through DrugBank, bringing together both company’s offerings to provide streamlined access to a broader range of high quality products.
The cost of artificially short expiration dates: Worsened shortages, higher costs and more waste As drug shortages have made headlines over the past few years, the FDA has announced the extension of expiration dates on a variety of drug products.
doi: 10.2210/rcsb_pdb/goodsell-gallery-048 The Virus that Cures It’s been over 25 years since the science magazine Discover first ran an extraordinary article about how a long-forgotten medical treatment, used in the former Soviet country of Georgia, could save us from the growing threat of untreatable, drug-resistant infections.
Responsible for “Protecting Human and Animal Health,” CVM makes sure that the drugs, devices, and food we provide for our pets are safe and effective. CVM does not regulate the practice of veterinary medicine (that’s a state licensing board) or vaccines for animal diseases (that’s USDA).
Food and Drug Administration (FDA) has determined that OAV-101 intrathecal (IT) clinical trials for spinal muscular atrophy (SMA) patients may proceed, thereby lifting the partial clinical trial hold initiated in October 2019. Novartis today announced that the U.S. SVP, Chief Medical Officer, Novartis Gene Therapies.
December has a huge number of legislative deadlines associated with the Food and Drug Omnibus Reform Act of 2022. Drug Importation: We may start the month of November with greater clarity about the FDA’s thinking about prescription drug importation plans.
Read Inhalable nanoparticles, packaged with mRNA or CRISPR systems, efficiently edit lung cells. Large-scale purification of functional AAV particles packaging the full genome using short-term ultracentrifugation with a zonal rotor. Nature Biotechnology. Gene Therapy. 📰 News A blood test to detect about 50 different cancers.
Food and Drug Administration (FDA) has approved CABENUVA (consisting of Janssen’s rilpivirine and ViiV Healthcare’s cabotegravir), the first and only once-monthly, long-acting regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults. TITUSVILLE, N.J. ,
WARNINGS AND PRECAUTIONS.
See “Worldwide Pro Forma Revenue” in Quarterly Package of Financial Information for this quarter, which is available on bms.com/investors/financial-reporting/quarterly-results, for information on the revenue of the company and Celgene on a stand-alone basis for the prior-year period. Food and Drug Administration (U.S. Regulatory.
The company announced donanemab received Breakthrough Therapy designation for treatment of Alzheimer’s disease and its intention to submit a biologics license application (BLA) for donanemab under the accelerated approval pathway later this year based on data from TRAILBLAZER-ALZ. Business Development/Other Developments.
The companies share a commitment to making the antibody cocktail available to COVID-19 patients around the globe and will support access in low- and lower-middle-income countries through drug donations to be made in partnership with public health organizations. None of the SAEs were considered to be related to study drug.
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). Pfizer Inc. NYSE: PFE) today announced that the U.S.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content