This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
In an exciting breakthrough, researchers have identified cancer drugs that might reverse the effects of Alzheimer's disease in the brain. By analyzing gene expression in brain cells, they discovered that some FDA-approved cancer medications could reverse damage caused by Alzheimer's.
as director of the Center for Drug Evaluation and Research (CDER).The WEDNESDAY, July 23, 2025 -- The U.S. Food and Drug Administration has appointed George Francis Tidmarsh, M.D., The CDER ensures the safety and effectiveness of.
Butler Recent reductions in force (RIFs) and leadership changes at FDA are already affecting key agency functionsand as the administration plans a broader reorganization, the impact will likely grow. Thats all bad news if your timeline depends on FDA sticking to theirs. By John W.M. Claud & Michelle L.
abscessus, through a virtual screening of FDA approved drugs on salicylate synthase. A drug repurposing strategy was applied against M. Eleven potential ligands were found; among these, three were confirmed as potent inhibitors of the enzyme.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
But growing ethical scrutiny, supply shortages, cost burdens, scientific innovation and regulatory shifts like the US Food and Drug Administration (FDA)’s new alternative methods roadmap are bringing the continued reliance on live NHPs into question, and opening the door to next-generation solutions that could eventually replace them altogether.
Chief Executive Officer Steve Herne has spent more than 25 years in clinical research, with senior roles at WCG, Bioclinica and Covance. The company worked closely with agencies including the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) from the outset, aligning its methodology with evolving guidance.
FDA Approves Streamlined Monitoring Requirements and REMS Program Removal for Bristol Myers Squibb’s CAR T Cell Therapies Breyanzi and Abecma, Marking Milestone Toward Expanding Access to Cancer Treatment In a significant regulatory development, Bristol Myers Squibb announced that the U.S.
Oncology drug approvals in H1 2025 In the first half of 2025, the FDA’s Center for Drug Evaluation and Research (CDER) approved a total of 16 novel drugs , with half of these drugs related to the treatment of cancer.
On April 10, 2025, the US FDA announced that it has a long-term plan to eliminate conventional animal testing in drug development, starting with monoclonal antibodies (mAbs).[ 2] An overview of the 3Rs The FDA and other global regulatory health authorities have long embraced the 3Rs of animal research (replace, reduce, and refine).
FDA Approves Label Update for Lilly’s Amyvid, Expanding Its Role in Alzheimer’s Disease Diagnosis and Therapy Guidance In a major development for Alzheimer’s diagnostics, Eli Lilly and Company (NYSE: LLY) announced that the U.S. At the time, its use was primarily restricted to supporting the diagnostic process.
The pharmaceutical industry has undergone significant changes over the past decade, with a growing trend towards outsourcing key aspects of research, development, and manufacturing to third-party vendors. The Rise of Integrated CDMOs The global biotechnology and pharmaceutical services outsourcing market size was valued at $70.48
Generative artificial intelligence (AI) has captured global attention for its transformative potential across industries, and nowhere is the promise greater — or more fraught — than in health care and clinical research. These domains are ripe for innovation. Errors jeopardize patient safety, regulatory compliance and trust.
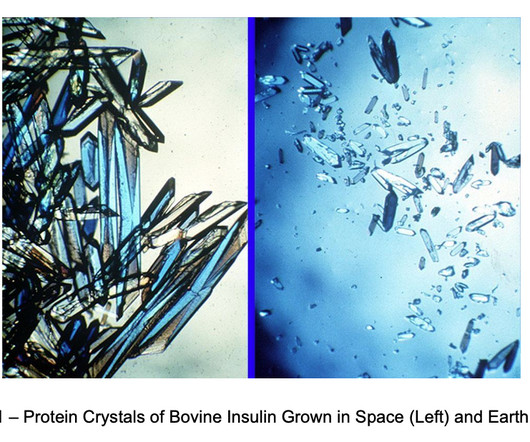
Merck, Bristol-Myers Squibb, Amgen, Eli Lilly, and Gilead have all conducted microgravity research in orbit. With respect to the latter, Mercks microgravity research on Keytruda is an informative case study. Despite the progress made, microgravity research projects to date have been exploratory, small-scale one-offs.
Food and Drug Administration (FDA)-approved inhibitors that broadly block FGFR. The work opens the door to potential targeted therapy for these tumors, particularly for pediatric low-grade gliomas (pLGG), though more research is needed to improve the efficacy of these treatments and to test them in pediatric clinical trials.
Leveraging the extensive breadth of available data to identify entities and relationships across data sources, clinical research experts, therapeutic specialists, machine learning (ML) engineers and others can collectively evaluate areas of interest that may create new opportunities for the asset and a broader clinical strategy.
While biomarkers are increasingly being adopted as surrogate endpoints by the US Food and Drug Administration (FDA), advances in biomarker detection also present a variety of regulatory challenges related to the complexities in their selection, implementation and clinical interpretation. References FDA-NIH Biomarker Working Group.
Two technologies, one goal: advancing precision oncology With a background in biochemistry and biology, Julien oversees Orano Med’s internal and external research programmes. By bringing our respective expertise together, we were able to screen and rapidly identify suitable candidates for RLT.”
Combination products are regulated by multiple divisions within the FDA, including: Drug/Device combination products are subject to the requirements of both the FDA’s Center for Drug Evaluation and Research (CDER) and the Center for Devices and Radiological Health (CDRH).
FDA Orphan Drug Designation for Antibody-Mediated Rejection in Solid Organ Transplantation The U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation (ODD) to riliprubart , an investigational immunology therapy developed by Sanofi, for the treatment of antibody-mediated rejection (AMR) in solid organ transplantation.
FDA Approves Merck’s ENFLONSIA™ to Protect Infants from Severe RSV Illness Merck operating as MSD outside the United States and Canada, has received a significant regulatory milestone with the U.S. Food and Drug Administration (FDA) granting approval for ENFLONSIA™ (clesrovimab-cfor). A New Era in Infant RSV Protection Dr. Dean Y.
There is no cure or effective treatment for this rare genetic disease, but new research suggests a potential path to one.& & Researchers at the Broad Institute and The Jackson Laboratory have used prime editing, a precise and versatile form of gene editing, to correct the root cause of AHC in mice.
The result of their research is what’s now described as targeted protein degradation, which conceptually is a bit like ordering a junk hauling service for the cell. For years, researchers struggled to develop an effective method for directing the E3 ligase. You can unsubscribe at anytime.
FDA Approves ANDEMBRY (garadacimab-gxii) The First Once-Monthly Prophylactic HAE Therapy Targeting Factor XIIa CSL a leading biotechnology company with a strong track record of developing innovative medicines for patients with rare and serious disorders, today announced that the U.S.
By: Juliane Mills, Senior Director, Therapeutic Strategy Lead, Rare Disease The rise of patient-led clinical research, particularly in rare disease, represents a significant shift in the clinical trial landscape. Why Is There an Increase in Patient-Led Rare Disease Research?
Xywav: A Low-Sodium Alternative with FDA Approval Xywav is a uniquely formulated, low-sodium oxybate therapy, and remains the only product of its kind approved by the U.S. Food and Drug Administration (FDA) for the treatment of cataplexy or excessive daytime sleepiness (EDS) in patients aged seven years and older with narcolepsy.
Researchers must characterize the anti-drug-antibody (ADA) response in preclinical and clinical studies and report any ADA-positive samples as a risk-based approach. The FDA's draft guidance on nonclinical safety assessment calls for tailored toxicology studies to address these potential risks.
These standards are widely recognized by regulatory agencies such as the FDA and EMA for use in analytical method development, validation, and quality control. Researchers can efficiently assess critical quality attributes such as vector integrity, purity, and size distribution throughout the development and manufacturing lifecycle.
This design was meant to enable clinician-researchers to gauge the ability of rilzabrutinib to control disease activity after the withdrawal of standard therapy. Fast Track and Orphan Drug Designations from FDA The U.S. Subsequently, rilzabrutinib was continued as monotherapy for the rest of the 52-week study period.
The interim data, presented at the SMA Research & Clinical Care Meeting hosted by Cure SMA in Anaheim, California, offer a strong signal of therapeutic potential. Under the terms of the agreement, Biogen holds the exclusive worldwide rights to develop, manufacture, and commercialize salanersen.
This was in 2006, at a time when the FDA guidances on these topics had not yet been published. Over time, this group of professionals evolved and grew, having regular stakeholder interactions with the FDA and Controlled Substances staff to discuss requirements for drug developers. corticosteroids, beta-blockers, antidepressants).
We are grateful to the patients, families, and researchers who made this progress possible, and we look forward to working with the health authorities to make treatment available as soon as possible.” Food and Drug Administration (FDA) and the European Medicines Agency (EMA) —to expedite review and potential approval for this new indication.
HK), a leading global Contract Research, Development, and Manufacturing Organization (CRDMO), has announced the official launch of WuXiHigh™ 2.0 , a next-generation, high-throughput formulation development platform specifically engineered for high-concentration biologics. WuXi Biologics Launches WuXiHigh™ 2.0 WuXiHigh™ 2.0:
19] Society and culture Legal status In October 2024, the US Food and Drug Administration (FDA) approved inavolisib for the treatment of PIK3CA -mutant breast cancer based on the results from the INAVO120 trial. [3] 3] [6] [20] [21] The drug application was granted priority review and breakthrough therapy designations by the FDA. [3]
Despite the potential efficiencies and the FDA encouraging manufacturers to make the switch from batch processing, the pharmaceutical industry has been slow to adopt continuous manufacturing due in large part to general conservatism and the cost implications of such investments. “Those are through the FDA,” he said.
In Part 2 of our conversation with Layla Hosseini-Gerami, Chief Data Science Officer at Ignota Labs , we explore the many forms of toxicity and how AI-powered tools like omics and cell painting are transforming early prediction in pre-clinical research.
I went on to complete my MBA and PhD at The Institute of Cancer Research (ICR) in drug development. This suited my personality and constant curiosity more than academic research. This pathway, called the PI3K pathway, had been largely researched and discarded. This has empowered me.
The Merkin Prize recognizes novel technologies that have improved human health and is administered by the Broad Institute, one of the worlds leading biomedical research institutes. Some researchers suspected that the body would attack and destroy the CAR T cells. Their work puts us on the doorstep of finding cures to help millions.
FDA Priority Review and Accelerated Development Pathway Sibeprenlimab has already drawn regulatory attention. Given the FDA’s acceptance of proteinuria reduction as a surrogate endpoint in IgAN trials, the strong Phase 3 results could support accelerated approval of sibeprenlimab pending final data.
Related groups Liu Group Related news New CRISPR genome editing system offers a wide range of versatility in human cells Researchers extend power of gene editing by developing a new class of DNA base editors In May 2025, researchers announced that K.J. This unprecedented feat required diagnosing K.J.’s
& The researchers systematically studied genes and proteins across the whole genome and proteome in lung tumors and paired noncancerous lung tissues from more than 400 patients from North America, Eastern Europe, and Asia, making it the largest “proteogenomic” study of any cancer to date.
Vamorolone was approved by the FDA in October 2023 for the management of DMD in patients ≥2 years of age. 19] The US Food and Drug Administration (FDA) approved vamorolone based on evidence from a single clinical trial of 121 boys with DMD who were 4 to <7 years of age. Food and Drug Administration (FDA). 65 (9): 737–743.
This partnership eliminates the lag and limitations of traditional market research, enabling dynamic feedback loops that power more effective engagement and smarter field execution. The KeyOps survey solution supports flexible quantitative research – from brief, on‑demand queries to in‑depth ATUs or AAUs – providing fast insights.
2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] The FDA has long supported development of non-opioid pain treatment. acting director of the FDA’s Center for Drug Evaluation and Research. under nitrogen.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content