This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
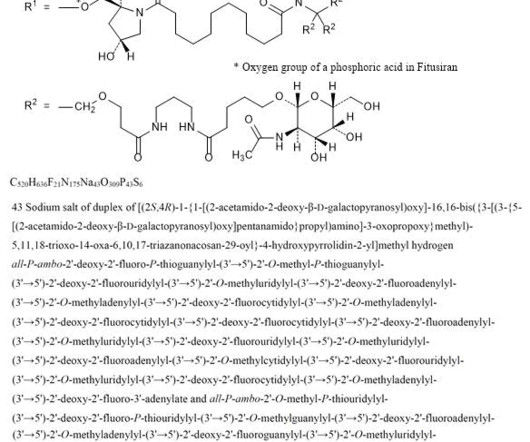
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 2] The label also has a warning about liver toxicity and the need to monitor liver blood tests at baseline and then monthly for at least six months after initiating treatment with fitusiran or after a dose increase of fitusiran. [2] Fitusiran 1711.0g/mol,
Dr Aaron Haubner, Senior Manager of North America Medical Affairs and Market Access at Terumo Blood and Cell Technologies , reveals that while promising new treatments emerge, urgent partnerships are needed to ensure this essential blood therapy reaches the patients who need it most.
4] It was approved for the treatment of major depressive disorder in the United States in September 2023. [4] 4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness. [5] 1] It is taken orally. [1] 1] It is taken orally. [1]
based subsidiary of Terumo and a global neurovascular company, announced today the FDAApproval of the PMA Supplement for the WEB 17 System, a new addition to the WEB Aneurysm Embolization System for the treatment of intracranial wide neck bifurcation aneurysms. The WEB System received its first PMA approval in late 2018.
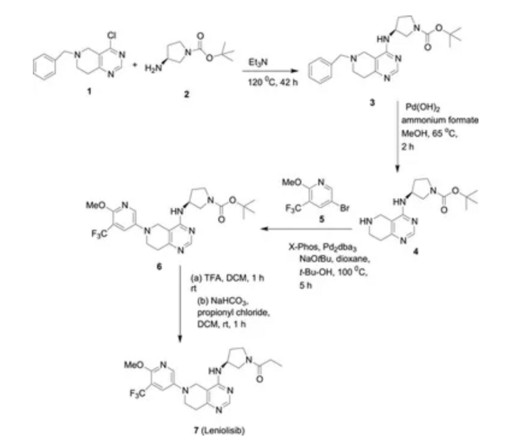
5] Leniolisib was approved for medical use in the United States in March 2023. [5] 5] [7] [8] It is the first approved medication for the treatment of activated PI3K delta syndrome. [5] 5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [9] World Health Organization (2016).
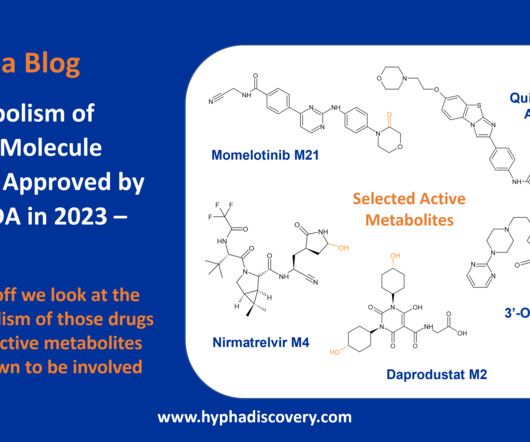
Metabolism of 2023 FDAApproved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. link] [3] FDA prescribing information for daprodustat. Basic Clin Pharmacol Toxicol. 131(5): 311- 324.
It is being investigated as a potential treatment for various herpes infections, including those resistant to traditional antivirals like acyclovir. Pritelivir (development codes AIC316 or BAY 57-1293 ) is a direct-acting antiviral drug in development for the treatment of herpes simplex virus infections (HSV).
We know from our research that the majority of men prefer an oral treatment to an injection,” Myovant CEO Lynn Seely said. The secondary endpoint for increasing lifespan for patients with metastatic prostate cancer came in at 74% still alive after treatment with Orgovyx compared to 75% of those on Lupron. .
MR : Chimeric antigen receptor T-cell (CAR-T) therapy is very effective in treating patients with B-cell lymphoma, leukemia, and multiple myeloma, where we have six FDA-approved drugs. However, these treatments will eventually fail for the majority of patients, so there is a strong need for better CAR therapies.
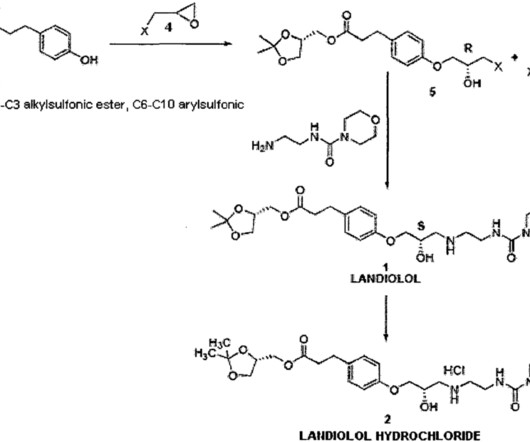
Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDAAPPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDAAPPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 9] It is used as landiolol hydrochloride.
As healthcare professionals, it’s our responsibility to educate patients about generic drugs and empower them to make informed decisions about their treatment options. Explain the FDAApproval Process Many patients are unaware of the rigorous approval process generic drugs must undergo. ” JAMA, 2016.
Rescheduling out of schedule I would allow for the medical use of FDA-approved prescription drugs dispensed by DEA-registered, state licensed pharmacies pursuant to prescriptions issued by similarly DEA-registered, state licensed practitioners. 12, 2016); Denial of Petition To Initiate Proceedings to Reschedule Marijuana, 81 Fed.
Food and Drug Administration (FDA) approval. Since then, the FDA has significantly changed its approach to rare and orphan diseases. The FDA Since 1983. The Orphan Drug Act of 1983 was instrumental in changing the number of orphan drugs approved in the U.S. Before 1983, only 38 orphan drugs had received U.S.
Food and Drug Administration (FDA) for the use of the oral anticoagulant Xarelto (rivaroxaban) in pediatric patients. There are currently no FDA-approved anticoagulation therapies for pediatric patients with congenital heart disease who have undergone the Fontan procedure. EINSTEIN-Jr.
June 8, 2021 /PRNewswire/ — Allergan Aesthetics, an AbbVie company (NYSE: ABBV ), is changing the way BOTOX ® Cosmetic, the #1 selling neurotoxin treatment 1 , is connecting with consumers. IRVINE, Calif. Experience the interactive Multimedia News Release here: [link]
. Each story is unique. Each story is unique.
Credit: Zhang & Chen, 2016, Cell 167, 1586–1597. Specifically, after 2 to 4 weeks of treatment with this combination of two correctors and a potentiator, such patients experienced about a 10 percentage point improvement in forced expiratory volume in 1 second (FEV1), which is a key measure of lung capacity.
Scott and colleagues focused on six genes that encode potential drug targets licensed or in development by GlaxoSmithKline for the treatment of obesity or diabetes. That’s critical considering that just 1 in 10 drug candidates entering human clinical trials successfully goes on to receive FDAapproval [5]. 2016 March 4. [5]
The approach might also be put to use for optimizing drug candidates or for discovering new uses for cancer drugs that have already been FDAapproved. While that remains a tremendous challenge, this new method could help to bring the goal of discovering many more precise and effective cancer treatments within better reach.
The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug. 8,128,929 was submitted on December 8, 2016 (Docket No.
Results reinforce well-established safety profile of Dupixent – the first ever biologic medicine for atopic dermatitis currently approved for patients as young 6 years old. Parents and caregivers are challenged to find safe and effective treatment options,” said John Reed, M.D., In 2016, the U.S.
Based on a technology developed by Broad Institute core member David Liu’s laboratory, the treatment is the first in a series of new medicines being tested to treat rare diseases by repairing patients’ particular genetic misspellings. Some of these treatments, like K.J.’s, Some of these treatments, like K.J.’s, Today, K.J.
Relevant here, FDA interpreted in Guidance that a proposed injectable biosimilar must “demonstrate that its product has the same strength as the reference product by demonstrating that both products have the same total content of drug substance (in mass or units of activity) and the same concentration of drug substance.”
Treatment induced sustainable clinical responses and reduced systemic inflammation. Daratumumab already is approved for the treatment of multiple myeloma. The company is developing its platform for the treatment of chronic, inflammation-mediated autoimmune diseases, and is initially focused on the treatment of RA.
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. The FDA will follow these procedures for both agency-initiated operations (e.g.,
.” Four years earlier, in 2019, Gray had become the first patient with sickle cell anemia — a genetic disorder that causes red blood cells to become sticky and rigid — to receive an experimental treatment using CRISPR genome editing. It was first characterized in 2016 by Feng Zhang’s lab.
Houck — Scheduling Criteria Under the Controlled Substances Act (“CSA”) Schedule I: • High potential for abuse; • No currently accepted medical use in treatment in the U.S.; Schedule III: • Potential for abuse less than drugs or substances in schedules I and II; • Currently accepted medical use in treatment in the U.S.; 21 U.S.C. §
4 For the treatment of rare genetic disorders especially, drugs with genetically supported targets are more than twice as likely to be approved 5 , thereby indicating genetics and genomics can empower companies to develop better drugs. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs.
FDA conducted the eight-factor scheduling analysis required by the CSA in 2016 and found that marijuana continued to meet the scheduling criteria for remaining in schedule I. 12, 2016); Denial of Petition to Initiate Proceedings to Reschedule Marijuana, 81 Fed. 53,688 (Aug. 53,767 (Aug. 21 U.S.C. §
Food and Drug Administration (FDA) has approved PREVNAR 20 (Pneumococcal 20-valent Conjugate Vaccine) for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older. Following today’s FDAapproval, the U.S.
Scheduling Under the CSA Substances in schedule I of the CSA have a high potential for abuse, no currently accepted medical use in treatment in the U.S., Those in schedule III have a potential for abuse less than schedule I or II substances, have a currently accepted use in treatment in the U.S., 21 U.S.C. § 21 U.S.C. § 53,688 (Aug.
21 December 2020 — AstraZeneca’s Tagrisso ( osimertinib ) has been approved in the US for the adjuvant treatment of adult patients with early-stage epidermal growth factor receptor-mutated (EGFRm) non-small cell lung cancer (NSCLC) after tumour resection with curative intent. 0.23; p<0.0001).
In early August, risdiplam, an orally available small molecule that binds to a stem-loop structure in the SMN2 pre-mRNA, received FDAapproval for the treatment of spinal muscular atrophy , a devastating genetic disease. At the time of that publication risdiplam was also in clinical trials for the treatment of SMA patients.
Food and Drug Administration (FDA) for the use of XARELTO ® (rivaroxaban) in pediatric patients. If approved, XARELTO ® will be the first and only oral Factor Xa inhibitor indicated in the U.S. EINSTEIN-Jr is the largest pediatric study completed to date for the treatment of VTE. for use in pediatric patients.
That company’s breast cancer treatment Trodelvy, which was granted accelerated approval from the U.S. billion acquisition of Germany-based Myr GmbH, a company focused on developing therapies for treatment of chronic hepatitis delta virus (HDV), the most severe form of viral hepatitis. Most Read Today. Source link.
Additionally, new data will be presented during the European Hematology Association (EHA) Virtual Congress from June 9-17 , including findings from the informCLL real-world prospective observational registry assessing how real-world treatment patterns align with National Comprehensive Cancer Network (NCCN ® )-recommended regimens for CLL/SLL.
Current treatment options in this age group are primarily topical steroids, which can be associated with safety risks and may impair growth when used long-term. In 2016, the FDA granted Breakthrough Therapy designation for Dupixent for the treatment of severe atopic dermatitis (in children aged 6 months to 11 years).
Title Type Date Cleared by OIRA Legal Deadline None Notable FDA Comment Periods Closing in November and December FDA comment periods are typically open for 30-60 days, unless they are extended. Within 180 days, FDA will release a report on the proceedings of the public meeting and any recommendations.
. ” However, studies over the last six decades have repeatedly affirmed that ultrasound is safe , at least when used in the short durations common to medical imaging and treatment. At least six types of CAR-T cells have already passed through clinical trials and garnered FDAapproval.
We wondered how given that HHS and the Drug Enforcement Administration (“DEA”) conducted eight-factor scheduling analyses in 2016, concluding that there was “no substantial evidence that marijuana should be removed from Schedule I.” 12, 2016) ; Denial of Petition to Initiate Proceedings to reschedule Marijuana, 81 Fed. 12, 2016).
Next-generation biomarker cytology test supports World Health Organization’s goal to eliminate cervical cancer, which is nearly 100 percent preventable with proper screening, vaccination and treatment. Basel, 16 September 2020 — Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced U.S. Publication of the full IMPACT study findings is pending.
If approved for this indication, we believe MYFEMBREE has the potential to redefine care for women with endometriosis as an effective, one pill, once-a-day treatment option.”. We look forward to potentially bringing this important new treatment option to women with endometriosis.”. In the U.S., About MYFEMBREE®.
12/29/2023 FDORA, Section 3202 Orphan Drug Program : FDA will convene a public meeting (or several) to solicit input from stakeholders regarding rare disease patient burdens, treatment options, side effects of treatments, etc.
As reported by POLITICO , “The decision — if allowed by the Supreme Court to take effect — would roll back actions the federal government has taken since 2016 to make the pills more accessible, including rules allowing online ordering, mail delivery, and pharmacy dispensing of the drugs. Read AgencyIQ’s analysis of that case here. ]
1] Gepotidacin was approved for medical use in the United States in March 2025. [1] 1] Gepotidacin was approved for medical use in the United States in March 2025. [1] Chemical synthesis The synthesis of gepotidacin has been described in two patents in 2008 and 2016 and comprises 11 steps (Fig. January 2016). 2) [11,12].
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.































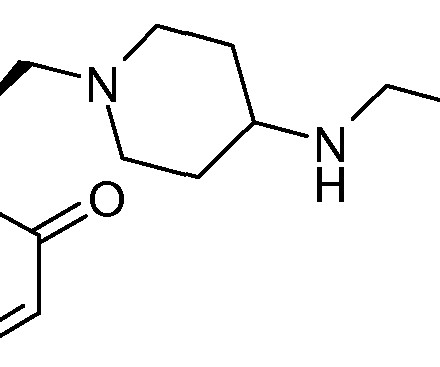






Let's personalize your content