This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Identifying branded drugs with a low likelihood of generic entry has become a crucial strategy for companies looking to expand their product portfolio through in-licensing. This approach not only helps maintain market exclusivity but also ensures a steady revenue stream for pharmaceutical companies.
This article was originally published by Ioana Gherghescu and Begoña Delgado-Charro in Pharmaceutics 2021, 13(1) under a Creative Commons Attribution License. Abstract Biosimilar medicines expand the biotherapeutic market and improve….
Biogen enters into a commercialization and license agreement to develop, manufacture and commercialize BAT1806, a proposed biosimilar referencing ACTEMRA ® (tocilizumab). Biosimilars have the potential to enable greater access to marketed biologic therapies while generating cost savings and healthcare sustainability.
Agreement includes commercializing biosimilar candidate of ophthalmology drug ranibizumab (Lucentis ® ) in Europe, Canada, Israel and global markets. Bioeq has in-licensed the exclusive global commercialization rights to FYB201 from the German biosimilar developer Formycon AG.
Click here to download a free report overview (including a summary of industry trends, the Table of Contents, and a List of Exhibits) New Drug Channels Institute Study: Biosimilars Are Delivering Higher Profits for Drug Distributors (press release) We’re offering special discounted pricing if you order before October 15, 2021! Section 4.4.
Drug Distribution Industry Expands as COVID-19 Disruption Fades and Biosimilars Boom (press release) We’re offering special discounted pricing if you order before October 21, 2022! We also update sections that were introduced in recent editions, such as our estimates of wholesalers' profits from provider-administered biosimilar drugs.
The FIE determination released in fall 2023 involves the Reference Product AbbVie’s Humira (adalimumab) and two interchangeable biosimilars—first approved is Cyltezo (adalimumab-adbm), filed by Boehringer Ingelheim Pharmaceuticals, Inc, which was followed by Abrilada (adalimumab-afzb), filed by Pfizer.
Updated guidance on promotional labeling for biosimilars and interchangeables emphasizes a similar approach Today, the FDA issued a revised draft guidance on the development of promotional labeling for biosimilars, reference products, and—newly—interchangeable products. regarding its administration, preparation, storage, or safety).
FDA’s new guidance on postapproval manufacturing changes for biosimilars focuses on current practice, new dosage forms Meeting a biosimilar user fee commitment, the FDA is expanding on its recommendations for biosimilar and interchangeable product applicants asking the FDA for post-approval manufacturing changes.
New FDA guidance on interchangeable biosimilar labeling heads to White House for review The FDA has submitted a draft guidance focused on the labeling of interchangeable biosimilar products to the White House for review, which would fulfill a Biosimilar User Fee Act (BsUFA III) commitment.
While that’s not to say that OTC hearing aids aren’t working to address the critical issue of hearing loss, it seems that almost two years is still not enough time to assess market impact, or to show that some of the promises of significant savings for large numbers of consumers have been realized.
This involves assessing the strength and breadth of patents, evaluating the potential for future patent challenges, and analyzing the value derived from licensing agreements and royalty streams. The high costs associated with bringing a new drug to market, estimated to be over $2.6
As always, we have updated all market and industry data with the most current information available, including our annual analyses of the market positions of the largest pharmacies, specialty pharmacies, and PBMs. If you have any questions before purchasing a license to the report, please email me. d/b/a Drug Channels Institute.
FDA recognizes that the FD&C Act exempts licensed healthcare practitioners from certain device regulations if they manufacture devices solely for use in the course of their professional practice. For example, FDA’s regulations have exempted from certain regulatory requirements (e.g.,
That clause excludes those ingredients that were first marketed as drug ingredients. MKP) asserting that “NMN is excluded from the dietary supplement definition under [FDC Act § 201(ff)(3)(B)(ii)] and may not be marketed as or in a dietary supplement.”
As always, we have updated all market and industry data with the most current information available. The report also updates our annual analyses of the strategies, market positions, and executive compensation of the three largest companies: AmerisourceBergen (Cencora), Cardinal Health, and McKesson. d/b/a Drug Channels Institute.
blood supply; and, Manufacturers of certain finished drug products marketed without an approved NDA or ANDA. The notification requirements apply to each individual manufacturer regardless of market share, the number of competitors making therapeutically equivalent products, or the amount of product in distribution.
About Toripalimab
Toripalimab was the first domestic anti-PD-1 monoclonal antibody approved for marketing in China. The FTD will significantly accelerate the research, development, and marketing of Toripalimab in the United States.
Claud — FDA’s Office of Pharmaceutical Quality (OPQ) in the Center for Drug Evaluation and Research (CDER) is charged with assuring that drugs marketed in the U.S. Of the 4,819 facilities in the Site Catalog, 60% manufacture drugs approved under a New Drug Application (NDA), Abbreviated NDA (ANDA), or Biologics License Application (BLA).
Under section 503B, drugs compounded by an FDA-registered outsourcing facility under the supervision of a licensed pharmacist can qualify for exemptions from FDA approval, labeling with adequate directions for use, and certain drug supply chain security requirements, subject to specific conditions. Draft Guidance III.B.2(e)
The length of time that it takes for us to complete clinical trials and obtain regulatory approval for product marketing has in the past varied and we expect similar variability in the future. We develop product candidates internally and through licensing collaborations, partnerships and joint ventures.
The FDA-required labeling is the drug labeling that is submitted by the sponsor and reviewed and approved by the FDA as part of the drug’s marketing application (including a New Drug Application, Abbreviated NDA, or a Biologics License Application) – and includes prescribing information (PI).
The length of time that it takes for us to complete clinical trials and obtain regulatory approval for product marketing has in the past varied and we expect similar variability in the future. We develop product candidates internally and through licensing collaborations, partnerships and joint ventures.
s MHRA unveiled details of its new International Recognition Procedure, which will allow the MHRA to rely on marketing authorizations by reference regulators from several countries for a wide range of products, including generics and those that received expedited review. market more quickly. The procedure is available for E.U.
Title Type Comments Close Enforcement Policy for Certain In Vitro Diagnostic Devices for Immediate Public Health Response in the Absence of a Declaration Under Section 564 Draft Guidance July 5 Consideration of Enforcement Policies for Tests During a Section 564 Declared Emergency Draft Guidance July 5 Financial Transparency and Efficiency of the Prescription (..)
While generic drugs technically pre-dated the law (look up “Paper NDAs” if you’re interested), the law turbocharged the ability of generics to come to market. We expect the FDA to mark the occasion, especially since drug pricing continues to be such a potent issue in the Presidential election. and the E.C.
The length of time that it takes for us to complete clinical trials and obtain regulatory approval for product marketing has in the past varied and we expect similar variability in the future. We develop product candidates internally and through licensing collaborations, partnerships and joint ventures.
FDA’s assumption that 50% of the tests will be exempt is particularly baffling because the laboratories are ones licensed to perform high complexity tests. 20, 2023) (“The lack of an equivalent IVD on the market is the primary reason labs develop LDTs.”) [7] PRIA. 8] FDA assumes that user fees will cover much of the increased costs.
If the confirmatory trial does not show that the drug provides clinical benefit, FDA has regulatory procedures in place that could lead to removing the drug from the market. 156 or case law that would support extension of the ‘929 patent that claims the product despite revocation of the biologics license application.
The length of time that it takes for Amgen to complete clinical trials and obtain regulatory approval for product marketing has in the past varied and Amgen expects similar variability in the future. Amgen develops product candidates internally and through licensing collaborations, partnerships and joint ventures.
Percent on an Operational Basis, Due to Biosimilar Competition; Global Skyrizi Net Revenues Were $1.590 Billion; Global Rinvoq Net Revenues Were $731 Million. percent on an operational basis, due to biosimilar competition. Humira Net Revenues Were $16.112 Billion, an Increase of 8.4 Percent on a Reported Basis, or 12.5 Recorded a $4.7
6/27/2023 Notification FDORA, Section 3201 Within 180 days of the passage of FDORA, all biologics and biosimilars sponsors must submit a written notice to the FDA of all actively marketed products (i.e., The following PDUFA dates were obtained from publicly available sources. not discontinued) and are available for sale.
Established ULTOMIRIS as new standard of care in PNH ahead of set goal, with more than 70% patient conversion from SOLIRIS ® (eculizumab) in 3 largest markets – U.S. ALXN2060 (AG10) – Eidos: Alexion holds an exclusive license to develop and commercialize ALXN2060 (AG10) in Japan. Received U.S. Germany & Japan.
The length of time that it takes for Amgen to complete clinical trials and obtain regulatory approval for product marketing has in the past varied and Amgen expects similar variability in the future. Amgen develops product candidates internally and through licensing collaborations, partnerships and joint ventures.
Shumsky — As readers of this blog know ( see, e.g. , here ), the Affordable Generics (and Biosimilars) Act has been floating around in Congress for the better part of two decades. The latest iteration of the Preserve Access to Affordable Generics and Biosimilars Act making its way through Congress is Senator Amy Klobuchar’s (D-MN) S.
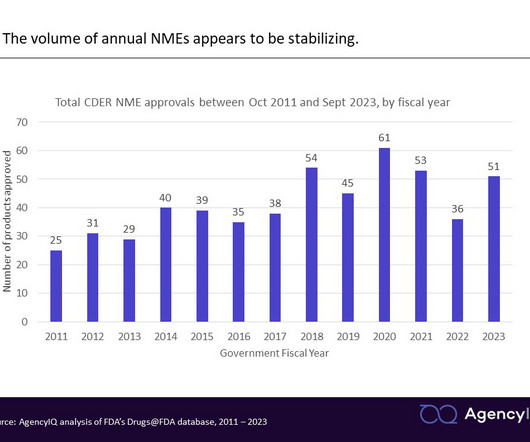
All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics). There were 21 NMEs added to the FDA’s Purple Book , which provides information about all biological products licensed by the FDA. Read the AgencyIQ analysis here.
Tokyo, Japan) today announced that the European Medicines Agency (EMA) has confirmed it has accepted for review, following a standard timetable, the Marketing Authorization Application (MAA) for aducanumab, an investigational treatment for Alzheimer’s disease. .
CAMBRIDGE, Mass. and TOKYO, Oct. Since October 2017 Biogen and Eisai Co.,
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. “An
MacroGenics , based in Rockville, Maryland, has a target action date of December 18 for its Biologics License Application (BLA) for margetuximab in combination with chemotherapy for patients with metastatic HER2+ breast cancer. Zai is strongly positioned to take advantage of a growing pharmaceutical market in this region.”.
As always, we have updated all market and industry data with the most current information available, including our annual analyses of the market positions of the largest pharmacies, specialty pharmacies, and PBMs. We also present a more unified outlook for specialty drugs, including specialty generics and biosimilars.
. – Second-quarter 2021 operating expenses increased 18 percent, driven primarily by higher research and development investments for late-stage assets, as well as higher relative marketing and selling expenses due to pandemic-related spending reductions in 2020. – Second-quarter 2021 earnings per share (EPS) decreased to $1.53
Seventeen products were orphan drugs, fourteen were generics and eight were biosimilars. Eight products received recommendations for conditional marketing authorization and one product was reviewed “under exceptional circumstances.” Reviews of orphan medicines and biosimilars have remained stable over the past three years.
After all, it never invested the time and resources necessary to obtain approval for commercial marketing or use. the marketing applicant before the Food and Drug Administration to support the application for patent term extension of U.S. That was the case in the PTO’s April 3, 1995 decision denying a PTE as to U.S. Lehman , No.
6/27/2023 Notification FDORA, Section 3201 Within 180 days of the passage of FDORA, all biologics and biosimilars sponsors must submit a written notice to the FDA of all actively marketed products (i.e., The following PDUFA dates were obtained from publicly available sources. not discontinued) and are available for sale.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content