This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This lets you focus on what matters most: driving meaningful results in drug research and development. From advancing drug discovery, managing clinical trials, or developing new healthcare solutions, reliable and flexible access to quality data is essential for success.
Introduction and background Singh: How do we bring scientists working in drug discovery into the world of AI? Recently, we’ve been pursuing AI product initiatives that have made me consider why people might be resistant to AI tools, what AI tools are capable of in drug discovery, and their limitations.
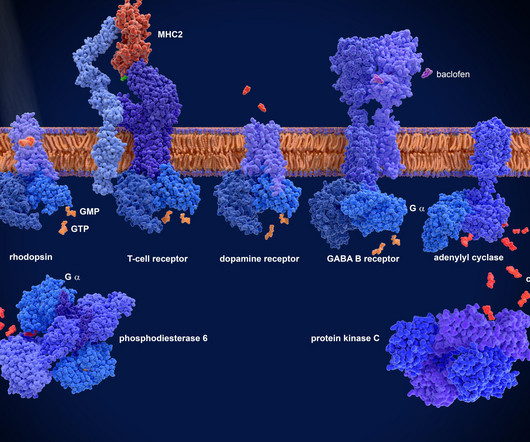
G protein-coupled receptors (GPCRs) are a highly validated drug target family – accounting for 30-35 percent of all approved drugs. Despite this, the therapeutic potential of targeting most GPCRs remains untapped, as only 10 percent of GPCRs have been drugged.
Developing treatments for individuals living with rare diseases is critical, but orphan drug development is laden with unique obstacles that necessitate innovative, multifaceted approaches. With the new EU HTA Regulation impacting orphan medicines, navigating these pathways has become even more challenging.
Additionally, the tablet form supports flexible dosing and simplifies dose reductions, which may be required due to drug interactions or adverse events, as guided by the product’s label. Food and Drug Administration (FDA) granted approval to the tablet formulation of BRUKINSA for all five of its approved indications.
Bringing a new therapeutic to market is a complex and multifaceted process culminating in submission and approval of a new drug application (NDA). Within the NDA, the chemistry, manufacturing, and controls (CMC) section plays a pivotal role in demonstrating drug quality, safety, and efficacy. Drug product (finished dosage form).
It also needed to be delivered directly to the liver cells in his body, rather than to cells in a lab, and this was achieved by packaging the base editor in a lipid nanoparticle, a delivery system conceptually similar to the one used to deliver COVID vaccines to billions of people. He needed a personalized, one-of-a-kind therapy.
The first of these components is the therapeutic genetic payload, or cargo, which is designed to correct or regulate the disease-causing mutation, whether that means offering a healthy copy of a mutated gene or delivering precision-editing tools like CRISPR. Still, AAV’s most obvious limitation remains its packaging capacity (~4.7
Navigating the complex landscape of drug development and manufacturing can be a daunting task. The Importance of Quality and Reliability In the high-stakes world of drug development, quality isn’t just important it’s everything. Assessing Your Technical Needs Every drug is unique, and so are its manufacturing requirements.
pyogenes protein — whose compactness makes them far easier to package into viral vectors and deliver into the human body. For example, one can fuse a deactivated Cas9 protein to so-called “transcriptional regulators” to either activate or repress genes without directly editing the genome at all.
Natural drugs, including delphinidin, which target the core genes, tripartite motif containing 16 (TRIM16), and protein regulator of cytokinesis 1 (PRC1), were then identified. ABSTRACT Pancreatic cancer (PC) is the leading cause of cancer-related death worldwide, and new biomarkers, therapeutic targets, and candidate drugs are needed.
Houck Drug Enforcement Administration (DEA) civil monetary settlements involving listed chemicals that do not include pseudoephedrine are rare. DEA seized five packages in Eagle Pass, Texas, identifying IMC Pro as the shipper containing 26.4 IMC Pro did not store or handle the packages nor maintain records about their contents.
They engineered genetically-encoded RNA exporters, based on viruses, that package and secrete RNA molecules in protective nanoparticles, allowing non-destructive monitoring of those RNA molecules in real-time. It demands protocols, regulations, and collaborative efforts between human beings. 7 Not to mention scaling things (i.e.
The Unsung Heroes of Generic Drugs: The Importance of Quality Assurance As we navigate the complex world of pharmaceuticals, it's easy to overlook the behind-the-scenes efforts that ensure the quality and safety of the medications we rely on. So, what can we learn from the importance of Quality Assurance in generic drugs?
However, as your high-school literature teacher warned youto ace the test, you need to read the book, ahem, source regulations, guidance, or other policy documents. The draft guidance recommends that no more than 15 questions are included in the briefing package. CBER will not commit to reviewing packages greater than 250 pages.
Food and Drug Administration (FDA) is already having ripple effects not just internally, but across the broader regulatory and life sciences communities and the public at large. FOIA is a U.S. In some cases, previously routine postings are delayed or going unpublished altogether.
Unlocking the Path to Affordable Medications: A Step-by-Step Guide to Generic Drug Approval As a healthcare professional or a patient advocate, you're likely no stranger to the importance of affordable medications. But have you ever wondered what it takes for a generic drug to hit the market?
D-ARABINOFURANOSYL)- 4′-C-Azido-2′-deoxy-2′-fluoro-b-D-arabinocytidine Azvudine is an antiviral drug which acts as a reverse transcriptase inhibitor. [3] In addition to its antiviral activity, concentration of the drug in the thymus has suggested immune-targeting activity; this dual function is unique among RdRp inhibitors.8
As a cornerstone of the drug development process, nonclinical investigational new drug (IND)-enabling studies are essential for supporting first-in-human (FIH) dosing for novel therapeutics. Pharmacology A typical IND-enabling package includes information on the primary, secondary, and safety pharmacology of the drug.
Originally developed in the 1970s to treat diabetes, these drugs—such as Ozempic, Wegovy, and Mounjaro—have become headline-makers for their ability to induce significant weight loss. Understanding GLP-1 drugs GLP-1, or glucagon-like peptide-1 receptor modulators, mimic natural hormones that regulate insulin and appetite.
FDA notes that it has, in the past, required single-unit or unit-dose containers for approved new drugs in ODT and film dosage form due to concerns about such products being more palatable than capsules or tablets. The suitable reference product must be a capsule or tablet that is swallowed.
What will the orphan drug market exclusivity haircut mean for industry? Just over a month ago, the European Commission released its proposal for the new pharmaceutical directive and regulation. Fill out the form to read the full article.
California publishes draft regulations for landmark plastic pollution reduction act CalRecycle has launched the formal rulemaking process for Senate Bill 54, which will implement a sweeping plan to reduce plastic waste in the state by 2032. By 2028, the state will require the recycling of 30% of single-use packaging and food service ware.
By Riëtte van Laack — FDA regulates pet food similar to other animal foods. The Federal Food, Drug, and Cosmetic Act (FDC Act) requires that all animal foods, like human foods, be safe to eat, produced under sanitary conditions, contain no harmful substances, and be truthfully labeled.
Artificial Intelligence (AI) is poised to transform the field of target discovery in drug development, offering immense potential to enhance efficacy, personalised medicine, and accelerate the development of innovative compounds. Cavlan notes that underpinning this use of AI in early discovery is the need to better understand biology.
This continued innovation highlights the complexity of the drug development process, particularly as the field is highly regulated by health authorities around the world. While it may not seem like a priority at first glance, the success of a drug can hinge on early decisions around artwork.
The Act is intended to address national security concerns by prohibiting certain conduct by regulated industry. In this regard we note that Medicaid is described in federal regulations as “Federal grants to States for medical assistance.” See, e.g., 42 CFR § 430.0.
The global ophthalmic drugs market size was valued at $33.81 Food and Drug Administration (FDA) published in March of 2022 pertaining to ophthalmic drugs and devices, titled Certain Ophthalmic Products: Policy Regarding Compliance With 21 CFR Part 4. Now, these companies are required to comply with new guidance from the U.S.
Of course, neither “cure” nor permanency are the bar for FDA to find that a new drug is effective; simply because there is the potential for something to be a “cure” or offer permanent/near-permanent effects does not mean it should be required to be such. That brings us to the product’s approval.
By Véronique Li, Senior Medical Device Regulation Expert & Ana Loloei & Allyson B. Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. Revised § 820.3
Determining if you have a combination product The high-level definition of combination product seems straightforward: a product with at least two constituent parts, such as: Drug/biologic (e.g., therapeutic drug/monoclonal antibody) Drug/device (e.g., insulin injector pen) Multiple drugs/device (e.g.,
What we expect European regulators to do in May 2024 In this recurring feature, AgencyIQ, through public data and previous analysis, determines what European medicine and device regulators will likely do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods, and more.
The authority to change drug labels outside of considerations for new safety information “could encourage third parties, such as academic investigators, insurance companies, and cooperative trial groups, to initiate such changes,” they wrote. .
One way to do so is to ensure the software platforms you are using to conduct research are in line with federal regulations. Computer systems used for clinical trials fall under Food and Drug Administration (FDA) 21 CFR Part 11. In 1997, FDA released regulations providing guidance on the use of electronic systems.
In this second of our two-part series, we continue our discussion about significant recent developments regarding the regulation of ophthalmic products and discuss what to expect in the months ahead. Food and Drug Administration’s (FDA) released updated guidance following the landmark Genus Medical Technologies LLC v. injectables).
Cannabis groups to Congress: FDA should regulate CBD as a dietary supplement U.S. legislators have asked for help to reimagine how the FDA should regulate cannabidiol (CBD) following the agency’s determination that it could not make use of its existing legislative or regulatory authorities to do so.
Kirschenbaum — The Calendar Year 2026 Medicare Physician Fee Schedule (PFS) proposed rule ( here ), which was issued yesterday by CMS, contained important amendments to the regulations on Medicare Part B average sales price (ASP) reporting. The payment limit for most Part B drugs is ASP plus 6 percent. with certain exceptions.
Jan Schakwosky has introduced the No Toxics in Food Packaging Act of 2023, which seeks to ban the use of ortho-phthalates, per- and polyfluoroalkyl substances, bisphenol compounds, styrene, and antimony trioxide as food contact substances. No Toxics in Food Packaging Act of 2023 On October 26, 2023, Reps. Key Documents and Dates H.R.
If the sponsor wants their drug approved, they need to complete all clinical studies and submit an application. Q: Please discuss the transfer of investigational new drug (IND) sponsorship from one sponsor to another and that process. Q: Can you ship a drug from another country to the U.S. A: Yes, the EUA is just temporary.
Baumhardt, Senior Medical Device Regulation Expert — The Unique Device Identification (UDI) System final rule requires all medical devices to bear a unique numeric or alphanumeric code in easily readable plain-text and machine-readable form. Devices can have both a UPC code and a UDI on their label and package.
However, an effective CRO doesn’t just perform assays; it provides comprehensive project planning, and also compiles, analyzes and packages data into an actionable and appropriate format for each individual client. Access to a laboratory partner that already possesses the equipment, knowledge and personnel can save time and money.
That’s terrific news for researchers looking for new ways to treat and prevent T2D, because it’s generally much easier to develop drugs that inhibit proteins than ones that boost their activity. ZnT8 is responsible for packaging zinc ions with insulin crystals, which are then secreted from the beta cells in the pancreas. 2014 Mar 2. [2]
government was put in place on February 27, 2023 and includes a number of proposed regulations to address trade relations between the E.U. First: all medicines for Northern Ireland must be approved by the MHRA The new regulation (EU) 2023/1182 applies to medicines that require marketing authorization. and the U.K.
What will the orphan drug market exclusivity haircut mean for industry? Just over a month ago, the European Commission released its proposal for the new pharmaceutical directive and regulation. Orphan designation in the E.U. provides a 10-year market exclusivity period. However, medical needs of patients are not sufficiently met.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content