This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Together, these technologies have entered at least 19 clinical trials, with clinical results from seven of those trials reported so far — all showing that the base edit or prime edit resulted in patient benefit. Of the 19 base editing and prime editing clinical trials underway now, more are taking place outside the U.S.
The treatment, now known as Casgevy, became the first CRISPR-based therapy to gain FDAapproval, in 2023. Dozens more clinical trials, based upon similar gene-editing technologies, are now underway. pyogenes protein — whose compactness makes them far easier to package into viral vectors and deliver into the human body.
Although Amodei does acknowledge some real-world issues limiting scientific progress — such as the slow growth of organisms and tedious clinical trials — he mostly passes over the more general tools that will be required to accelerate research in the near term. This essay focuses on how we might do both, specifically for the cell.
Explain the FDAApproval Process Many patients are unaware of the rigorous approval process generic drugs must undergo. Educate them about the FDA’s role in ensuring the safety and efficacy of generic medications. These anecdotes can help alleviate fears and build confidence in generic medications.
Limited Evidence for Nalfmefene "In 2021, due to the widespread availability of high-potency synthetic opioids like fentanyl, the US FDAapproved two high-dose naloxone products, an 8 mg IN spray (Kloxxado) and a 5 mg IM injectable (Zimhi). In 2023, the FDAapproved a 2.7
Biederman, Director of Cardiovascular Imaging and Cardiac MRI at Allegheny General Hospital/Allegheny Health System in Pittsburgh and an author on the groundbreaking 2017 MagnaSafe trial published in NEJM. approval complements previous MR-conditional approvals in geographies outside of the United States.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
Dive into this week’s update for more details on the actions taken by the FDA in the ongoing response to the Covid-19 pandemic. FDAapproves first treatment for Covid-19. On October 22, the FDAapproved the antiviral drug Veklury for use in adult and pediatric patients for the treatment of Covid-19 requiring hospitalization.
The authority to change drug labels outside of considerations for new safety information “could encourage third parties, such as academic investigators, insurance companies, and cooperative trial groups, to initiate such changes,” they wrote. .
Clinical trials for ultra-rare diseases can be particularly challenging to mount due to small, geographically-dispersed patient populations. For such trials, the US Food and Drug Administration (FDA) may allow the use of credible real-world data (RWD) and real-world evidence (RWE) in lieu of data collected in a Phase 3 trial.
The sponsor is the pharmaceutical company conducting the trial. If you mean using a different contract research organization (CRO) for the different phases of clinical trials – that’s different. Also consider CRO oversight, trial management, data handling and record keeping, as well as allocation of responsibilities.
Today’s data, involving an additional 524 patients from the ongoing Phase 2/3 trial, provides definitive final virology results and meets the clinical endpoint of reducing medical visits. REGN-COV2 was well tolerated in the trial. TARRYTOWN, N.Y. , 28, 2020 /PRNewswire/ — . Regeneron has shared these results with the U.S.
However, when it comes to an IND and supporting a clinical trial, FDA’s primary focus is on healthy volunteer and patient safety. It is critical that the nonclinical program outlined in the PIND briefing document is presented in a manner that allows FDA to provide relevant input on the required IND-enabling studies.
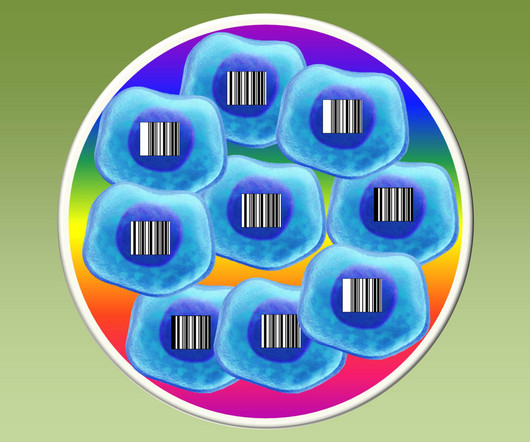
Instead of the black, printed stripes of the Universal Product Codes (UPCs) that we see on everything from package deliveries to clothing tags, they used short, unique snippets of DNA to label cells. In their trial run of PRISM, the researchers uniquely barcoded more than 100 cancer cell lines.
30, 2020 /PRNewswire/ — The IDMC also recommends continuation of enrollment in the REGN-COV2 outpatient trial. NASDAQ: REGN) received today a recommendation from the independent data monitoring committee (IDMC) for the REGN-COV2 antibody cocktail treatment trials for COVID-19 that the current hospitalized patient trial be modified.
Silverback’s lead candidate is currently in a Phase I trial in adults with HER2-expressing solid tumors. BioAge is on the cusp of taking pilot therapies BGE-117 and BGE-175 into clinical trials, targeting the first half of 2021. Pear’s reSET, reSET-O and Somryst are the first PDTs to receive FDAapproval for treating disease.
1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2. 2 Since then, the FDA has approved four more AAV-based gene therapies—Zolgensma, Hemgenix, Elevidys and Rocktavian—for treating various diseases.
Injectable semaglutide 1 mg is a GLP-1 receptor agonist and the highest dose of injectable semaglutide FDA-approved for the treatment of type 2 diabetes. SURPASS-2 was a 40-week, randomized, open-label trial comparing the efficacy and safety of tirzepatide to semaglutide as an add-on to metformin in adults with type 2 diabetes.
NASDAQ: REGN ) today announced that the New England Journal of Medicine (NEJM) has published initial clinical data from an ongoing seamless Phase 1/2/3 trial of the antibody cocktail casirivimab and imdevimab in non-hospitalized patients with COVID-19. TARRYTOWN, N.Y. , 17, 2020 /PRNewswire/ — Regeneron Pharmaceuticals, Inc.
Potentially registrational Phase 2 portion of the trial has been initiated and is enrolling patients. NASDAQ: REGN) today announced updated data for REGN5458, a BCMAxCD3 bispecific antibody, from the Phase 1 portion of a Phase 1/2 trial in patients with relapsed or refractory (R/R) multiple myeloma. Regeneron Pharmaceuticals, Inc.
CABENUVA, a co-packaged kit with two injectable medicines, offers people living with HIV a new approach for maintaining viral suppression. Click to Tweet : #BREAKING: The #FDA has approved another treatment option for people living with #HIV. Global Head, Janssen Research & Development, Johnson & Johnson.
A significant effort has been made to correctly map the drug form of the EMA data by manually inspecting different EMA sources of information, such as the Product Information (Annex I: Summary of Product Characteristics and Annex III: Labelling and Package Leaflet) and/or Assessment Report, where available. University of Dundee: T.
As in earlier outpatient trial, immune status when patients entered the trial was a strong predictor of viral load and clinical outcomes. The primary clinical objective of this initial analysis was to determine if there was sufficient efficacy in these patients to warrant continuing the trial (i.e., futility analysis).
“Patients in our antibody cocktail outpatient clinical trial experienced significant reductions in virus levels and required fewer medical visits for COVID-19, suggesting the therapy can help reduce the current burden on hospitals and healthcare systems,” said George D. .”
To this point, Moderna has only submitted two months of follow-up safety data, and the FDA typically requires six months for a full approval. intends to ship just shy of six million doses of Moderna’s vaccine once the FDAapproves EUA. Initial doses are expected to be limited as manufacturing ramps up.
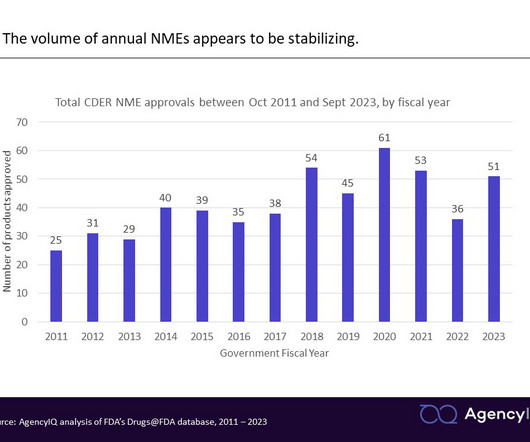
While the definition of NME has changed over the years, it can sometimes include a combination product consisting of at least one drug that has previously been approved. Data on these novel approvals is published throughout the year by both CDER and CBER. FDAapproved 13 NMEs through the AA pathway in FY 2023, making up 25.5%
The vaccine was shown in clinical trials to be safe and effective at preventing symptomatic COVID-19, with no severe cases and no hospitalisations more than 14 days after the second dose. The approval in India is an important milestone as it will enable to supply India but also a large number of countries around the world.
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Under the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 (PAHPRA) , the FDA also has some authority to extend MCM expiration dates.
The clinical evidence from Regeneron’s outpatient trial suggests that monoclonal antibodies such as casirivimab and imdevimab have the greatest benefit when given early after diagnosis and in patients who have not yet mounted their own immune response or who have high viral load.
See “Worldwide Pro Forma Revenue” in Quarterly Package of Financial Information for this quarter, which is available on bms.com/investors/financial-reporting/quarterly-results, for information on the revenue of the company and Celgene on a stand-alone basis for the prior-year period. Oncology and Hematology. Regulatory. Regulatory.
REGN-COV2 trial in the COVID-19 outpatient setting met primary and key secondary endpoints. FDA accepted for priority review Libtayo ® (cemiplimab-rwlc) for both advanced non-small cell lung cancer and basal cell carcinoma. FDAapproved Inmazeb for Ebola ( Zaire ebolavirus). and non-GAAP diluted EPS (1) was $8.36 .
I want to thank the thousands of participants in our Phase 1, Phase 2 and Phase 3 studies, as well as the staff at clinical trial sites who have been on the front lines of the fight against the virus. The trial will continue to accrue additional data relevant to safety and efficacy even after an EUA is submitted. ages 18 and older.
Read Inhalable nanoparticles, packaged with mRNA or CRISPR systems, efficiently edit lung cells. Large-scale purification of functional AAV particles packaging the full genome using short-term ultracentrifugation with a zonal rotor. ” Clinical trials are underway. Nature Biotechnology. The Lancet HIV. Gene Therapy.
12/29/2023 FDORA, Section 3602 Clinical Trials Modernization : FDA is directed to require the submission of a “diversity action plan” for all Phase 3 clinical trials of new drugs. FDA is directed to issue new draft guidance or update existing guidance regarding Diversity Action Plans for clinical studies.
The clinical evidence from Regeneron’s outpatient trial suggests that monoclonal antibodies such as REGEN-COV2 have the greatest benefit when given early after diagnosis and in patients who have not yet mounted their own immune response or who have high viral load. Data from these trials will be used to support a future BLA submission.
The FDA CRL cited inspection findings that arose from a “multi-sponsor inspection of a third-party, contract manufacturing organization” which involved the monoclonal antibody drug substance for lebrikizumab. The clinical package, safety and label were not affected. However, BeiGene announced that it is seeking approval in the U.S.
Food and Drug Administration (FDA) approved LUMAKRAS for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic NSCLC, as determined by an FDA-approved test, who have received at least one prior systemic therapy. In May, the U.S. AMG 451 / KHK4083. Manufacturing Facilities.
Most people would take the two CRISPR gene-editing components (a Cas9 protein and guide RNA), package them up inside of a virus, and then inject the viruses into the skulls of mice. Read Ghana is the first country to approve a new malaria vaccine developed at the University of Oxford. Stahl et al. on bioRxiv.
Most people would take the two CRISPR gene-editing components (a Cas9 protein and guide RNA), package them up inside of a virus, and then inject the viruses into the skulls of mice. Read Ghana is the first country to approve a new malaria vaccine developed at the University of Oxford. Stahl et al. on bioRxiv.
In the case of semaglutide, those cells are Saccharomyces cerevisiae— also known as Baker’s yeast — engineered to secrete a peptide precursor that is later purified, chemically modified, packaged into an injectable or tablet form, and then shipped around the world. Continuing this method may not scale.
CDMO Services assumes continued growth in Development Services (DVS), Drug Substance (DS) manufacturing, and Drug Product (DP) manufacturing and Packaging for both clinical- and commercial-stage projects on behalf of a growing list of pharmaceutical and biotechnology innovators and government/NGO customers.
FOOTNOTES. (1)
BY LAURA DIANGELO, MPH | MAR 6, 2024 5:54 PM CST Fiscal year 2024 appropriations bills On March 3, 2024, House and Senate appropriators released a package of final fiscal year (FY) 2024 appropriations bills. The budget agreement “asks” the FDA for some information and activities, and “recognizes” some FDA authority in other areas.
The prebiotic may either feed the probiotic that it is packaged with, or it may feed another beneficial microbe that is intended to complement the activity of the probiotic. In fact, the FDA has even developed a guidance document for “early clinical trials with live biotherapeutic products [LBPs].”
Food and Drug Administration (FDA) approved Inmazeb (atoltivimab, maftivimab and odesivimab-ebgn) for the treatment of infection caused by Zaire ebolavirus in adult and pediatric patients, including newborns of mothers who have tested positive for the infection. TARRYTOWN, N.Y., NASDAQ: REGN) announced today that the U.S.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content