

The rising impact of biomarkers in early clinical development
Drug Target Review
APRIL 7, 2025
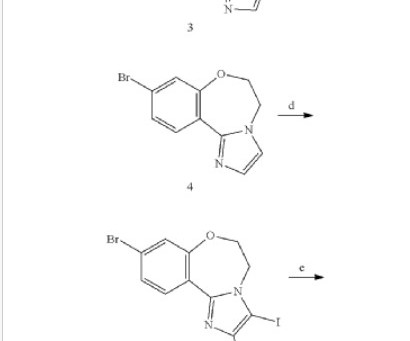
A surrogate endpoint is a marker used in clinical trials as a substitute for a direct clinical outcome. For example, transcriptomic processes are showing the potential to identify and track failures in gene expression and gene regulation of amyloid and tau-related biomarkers, understood as precursors to the onset of Alzheimers disease (AD).










































Let's personalize your content