This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Currently, more than 20% of FDA-approved monoclonal antibodies (mAbs) fall into this category, reflecting a growing industry trend. High viscosity , aggregation risks , and stability issues often complicate both upstream and downstream processes, including manufacturing, packaging, and storage. WuXiHigh™ 2.0: WuXiHigh™ 2.0:
Syn EuropeanJournalofMedicinalChemistry265(2024)116124 Vamorolone (Agamree) On October 26, 2023, Vamorolone, developed jointly by Santhera Pharmaceuticals and ReveraGen BioPharma, has received FDAapproval to treat DMD in patients aged 2 years and older [1]. Vamorolone: first approval. Drugs 2024, 84, 111− 117. (71)
s specific mutation, creating a mouse model of the disease, determining the optimal base editor, performing extensive safety analyses, working with Danaher to manufacture the therapeutic, conducting toxicity studies, and securing FDAapproval for the trial. This unprecedented feat required diagnosing K.J.’s
Casgevy , the first FDA-approved CRISPR-based therapy, is also an ex vivo gene therapy; it aims to cure sickle cell disease by editing a patient’s blood-producing stem cells so they generate functional fetal hemoglobin, then returning these corrected cells to the bloodstream.
The treatment, now known as Casgevy, became the first CRISPR-based therapy to gain FDAapproval, in 2023. pyogenes protein — whose compactness makes them far easier to package into viral vectors and deliver into the human body. Dozens more clinical trials, based upon similar gene-editing technologies, are now underway.
They engineered genetically-encoded RNA exporters, based on viruses, that package and secrete RNA molecules in protective nanoparticles, allowing non-destructive monitoring of those RNA molecules in real-time. Today, a single injection of an FDA-approved gene therapy, called Hemgenix , cures this disease.
Generic drugs play a crucial role in providing affordable medication options to patients. As healthcare professionals, it’s our responsibility to educate patients about generic drugs and empower them to make informed decisions about their treatment options. What Are Generic Drugs?
Limited Evidence for Nalfmefene "In 2021, due to the widespread availability of high-potency synthetic opioids like fentanyl, the US FDAapproved two high-dose naloxone products, an 8 mg IN spray (Kloxxado) and a 5 mg IM injectable (Zimhi). In 2023, the FDAapproved a 2.7 Int J Drug Policy. doi:10.1016/j.drugpo.2024.104587
To increase demand for products during the novel coronavirus pandemic, the US Food and Drug Administration (FDA) has already approved at-home testing and investigated the effectiveness of malaria pills and other antiviral drugs to treat Covid-19 9. FDAapproves first treatment for Covid-19. Source link.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
The authority to change drug labels outside of considerations for new safety information “could encourage third parties, such as academic investigators, insurance companies, and cooperative trial groups, to initiate such changes,” they wrote. . Posted 17 May 2021 | By Jeff Craven .
Effect on FDAapprovals— In cases where a biological product or drug needs to change aspects of its manufacturing processes to avoid using a covered equipment or service, will it need to file supplements with FDA for the CMC update?
The 505(b)(2) new drug application (NDA) pathway offers a unique opportunity for small molecule developers to bring innovative products to market more efficiently by leveraging existing data they do not own or have right of reference to. The overall development strategy for GT123 was to include a complete CMC package. Human factors.
When returned directly to participants, aggregate results should be packaged and communicated in user-friendly formats, such as in newsletters or web pages constructed with readability and health literacy principles in mind. Conversely, a CGM not yet FDA-approved assessed in the study for reliability, would yield an investigational result.
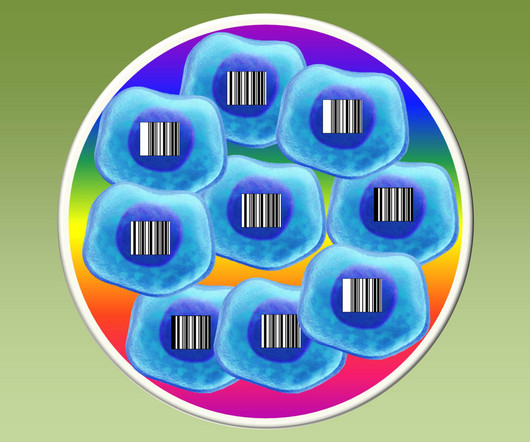
Instead of the black, printed stripes of the Universal Product Codes (UPCs) that we see on everything from package deliveries to clothing tags, they used short, unique snippets of DNA to label cells. In the next key test, the researchers exposed a mixture of 25 barcoded lung cancer cell lines to approved cancer drugs.
Farquhar — A drug manufacturer’s bad post-inspection grade from the U.S. Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDAapproval of applications to market drugs manufactured at the facility.
For such trials, the US Food and Drug Administration (FDA) may allow the use of credible real-world data (RWD) and real-world evidence (RWE) in lieu of data collected in a Phase 3 trial. The FDA also granted a second meeting for review.
Q: If an emergency use authorization (EUA) is granted, once there is an approved treatment, does that mean that the EUA is no longer valid? If the sponsor wants their drugapproved, they need to complete all clinical studies and submit an application. Q: Can you ship a drug from another country to the U.S.
Altasciences Chosen by Virpax to Support the Development of a New Drug to Prevent Spread of Flu-like Viruses pmjackson Wed, 09/20/2023 - 13:48 Laval, Québec, September 21, 2023 - Altasciences is pleased to have been chosen by Virpax Pharmaceuticals, Inc. Altasciences helps sponsors get better drugs to the people who need them, faster.
By William Salminen , Madelyn Huang, & Andrew Emanuel Without concrete guidelines, it can be confusing when determining what nonclinical studies are needed for a Pre-Investigational New Drug application (PIND) meeting. neurotoxicity) that require special assessments.
Vanda requested that FDA revoke the approval of Apotex’s and Teva’s generic versions of Hetlioz on the grounds that the generic tasimelteon products did not meet the statutory “same labeling” requirement for generic drugs found in 21 U.S.C. § patients in harm’s way.” FDA responded in late July, denying Vanda’s petitions.
As part of the comprehensive submission package to the European Marketing Authorization, Sandoz conducted a Phase I pharmacokinetics (PK) bridging study comparing its approved adalimumab 50 mg/mL 2 with the 100 mg/mL (HCF). The adalimumab reference medicine (Humira ® *) was first approved with an adalimumab concentration of 50 mg/mL.
AbCellera’s AI-powered antibody discovery platform speeds the otherwise lengthy and grueling process by analyzing the database of natural immune systems to find antibodies that can be developed into drugs. Tackle the toughest problems in drug development.” The promise to partners is to “move quickly. Reduce cost. Remix Therapeutics .
1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2. 2 Since then, the FDA has approved four more AAV-based gene therapies—Zolgensma, Hemgenix, Elevidys and Rocktavian—for treating various diseases.
FDA finalizes guidance on electronic submissions for OTC products FDA has fulfilled its commitment under the Over-the-Counter Monograph Drug User Fee (OMUFA) program to issue final guidance on how sponsors can electronically submit monographs and other documents. Read AgencyIQ’s explainer on the CARES Act here.
Food and Drug Administration , which is currently evaluating REGN-COV2 for a potential Emergency Use Authorization in mild-to-moderate outpatients at high risk for poor outcomes. Finally, the IDMC recommends continuation of the outpatient trial without modification. Regeneron is also informing the U.S.
Food and Drug Administration (FDA) for the treatment of mild to moderate COVID-19 in patients 12 years of age and older and weighing at least 40 kg, who have received positive results of direct SARS-CoV-2 viral testing and are at high risk for progressing to severe COVID-19 and/or hospitalization.
About the Phase 1/2 Dose-escalation Trial REGN5458 monotherapy is being investigated in an open-label, Phase 1/2 dose-escalation trial in patients with R/R multiple myeloma who are at least triple refractory to existing therapeutic options, including proteasome inhibitors, immunomodulatory drugs and CD38 antibody treatments.
“The first job of an antiviral therapeutic drug is to lower the viral load, and our initial data in 275 patients strongly suggested that the REGN-COV2 antibody cocktail could lower viral load and thereby potentially improve clinical outcomes. . Regeneron has shared these results with the U.S.
The past few years have seen multiple high-profile FDAapprovals for AAV-based therapeutics, with a series of regulatory decisions in the pipeline for 2025. This results in more fully packaged and intact capsids, which are essential for reducing manufacturing costs per dose and enhancing the effectiveness of gene therapies.
Food and Drug Administration (FDA) has approved CABENUVA (consisting of Janssen’s rilpivirine and ViiV Healthcare’s cabotegravir), the first and only once-monthly, long-acting regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults. TITUSVILLE, N.J. ,
We are delighted to announce the release of ChEMBL 34, which includes a full update to drug and clinical candidate drug data. 71 out of the 882 newly added EMA drugs are only authorised by EMA, rather than from other regulatory bodies e.g. FDA. This data is currently available for European Medicines Agency drugs only.
The product is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. After you fill in your information and confirm your package will be shipped for free right to your doorstep as soon as possible. Dentitox Pro is non-GMO and safe.
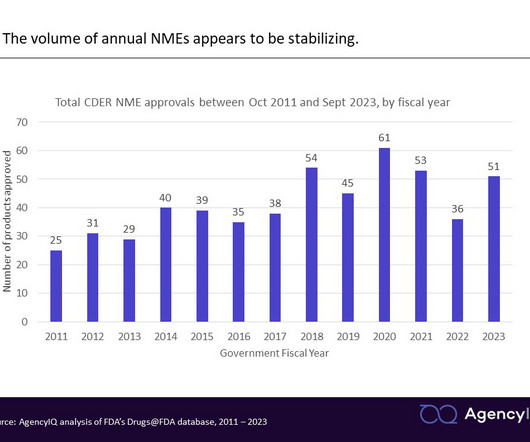
FDA fiscal year in review: New drugapprovals in the wake of the pandemic and legislative reforms AgencyIQ analyzed CDER’s novel drugapprovals in Fiscal Year 2023, identifying a recovery in approval numbers as the agency resumes a new normal following the pandemic. As a reminder, the U.S.
The cost of artificially short expiration dates: Worsened shortages, higher costs and more waste As drug shortages have made headlines over the past few years, the FDA has announced the extension of expiration dates on a variety of drug products.
Every capsule is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. If you order the 3 bottles or 6 bottles package (which we highly recommend as we estimate that we will run out of stocks soon as this has happened before) you’ll also take advantage of a huge discount.
Gaulkin — In May 2023, we posted about a CMS proposed regulation that sought to make a wide variety of changes to the Medicaid Drug Rebate Program (MDRP), including a new “price verification survey,” and a controversial proposal to require “stacking” of discounts to different customers when determining best price. Kirschenbaum & Sophia R.
Food and Drug Administration (FDA) advisory panel in regard to the COVID-19 vaccine from Moderna. To this point, Moderna has only submitted two months of follow-up safety data, and the FDA typically requires six months for a full approval. Boumen Japet/Shutterstock.
Perhaps the most important addition to FDA’s agenda is a proposed rule intended to clarify that Laboratory Developed Tests (LDTs) are to be regulated as medical devices under the Federal Food, Drug and Cosmetic Act (FD&C Act). Read our analysis of that rule here and here. ]
The companies share a commitment to making the antibody cocktail available to COVID-19 patients around the globe and will support access in low- and lower-middle-income countries through drug donations to be made in partnership with public health organizations. Casirivimab and imdevimab injection is not FDAapproved for any use.
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. Pfizer Inc.
December has a huge number of legislative deadlines associated with the Food and Drug Omnibus Reform Act of 2022. Drug Importation: We may start the month of November with greater clarity about the FDA’s thinking about prescription drug importation plans.
. | NOV 8, 2023 10:02 PM CST Regulatory background The Federal Food, Drug, and Cosmetics Act (FD&C Act) is administered by the Food and Drug Administration (FDA) and regulates cosmetics. MoCRA also requires all marketed cosmetic products to be listed with the FDA by December 29, 2023. less than $1 million.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content